介绍
B细胞转化可以发生在B细胞发育的不同阶段,分子分析表明B细胞恶性肿瘤具有极强的异质性。套细胞淋巴瘤(MCL)2是生发前中心非霍奇金淋巴瘤(NHL)的一个亚型,其特征是在几乎100%的病例中通过荧光检测到易位t(11;14)(q13;q32)就地杂交,将循环蛋白D1(CCND-1型)14q32免疫球蛋白重链位点11q13基因(1–三). MCL预后不良,被认为是不可治愈的(4). 同样,弥漫性大B细胞淋巴瘤(DLBCL)是世界上最常见的非霍奇金淋巴瘤,根据全球基因表达谱可分为三种不同类型:生发中心B细胞(GCB)-DLBCL、活化类B细胞(ABC)-DLBCL和原发性纵隔B细胞淋巴瘤(5,6). 在DLBCL中发现了不同的染色体易位,包括BCL2和BCL6重排,它们相互排斥(7–9). 尽管有这些遗传变异,但淋巴瘤细胞类型仍有一些共同特征。例如,我们之前的研究表明,蛋白质精氨酸甲基转移酶5(PRMT5)在不同类型的B细胞淋巴瘤中高度表达,包括MCL、GCB-DLBCL和ABC-DLBCL(10–12)并且参与B细胞永生化和恶性表型的维持(12,13). 这些发现支持了这样一种观点,即PRMT5通过调节保守的生长调节通路来控制淋巴瘤细胞的增殖和存活。
组蛋白甲基化是一种重要的表观遗传修饰,控制染色质结构和基因表达,并与癌症的发展有关(14–16). 然而,关于表观遗传修饰的作用及其对NHL发病机制的潜在影响的知识有限。虽然组蛋白赖氨酸甲基化的作用已被广泛研究,但组蛋白精氨酸甲基化仍大部分未被探索。PRMT5是一种II型精氨酸甲基转移酶,可以靶向组蛋白H3和H4并显著改变基因表达程序以促进细胞转化(10,12,13,17). PRMT5还能够甲基化非组蛋白,如p53和E2F1,这些修饰的直接结果是这些转录因子调节的特定靶基因的异常表达,伴随着癌细胞生长的增强(18,19). 我们之前已经证明PRMT5在NHL细胞中选择性过度表达,并且它是促进淋巴瘤细胞生长的主要生存因子(10). 我们还从机理上证明,PRMT5通过负调控RB/E2F抑癌途径促进淋巴瘤细胞生长和存活(11). 在后续研究中,我们证明了PRMT5通过使用双途径灭活RB/E2F通路促进多梳阻遏物复合物2(PRC2)的表达,该双途径包括通过CYCLIN D1-CDK4/CDK6使RB1过度磷酸化和PRMT5介导的RBL2表达的直接表观遗传沉默(12). 尽管PRMT5和RB-E2F信号通路之间的关系已经建立,但我们仍然不知道PRMT5控制促生存因子CYCLIN D1、c-MYC和SURVIVIN表达的机制。因为这三个基因都是真诚地β-CATENIN靶点,我们重点研究了PRMT5促进WNT/β-CATEN增殖信号传导的分子机制。
WNT(Wingless related integration site)信号通路在进化上是保守的,可以由大量WNT蛋白触发,在增殖、分化和发育过程中发挥重要作用(20–26). WNTs是富含半胱氨酸的分泌糖蛋白,可激活受体介导的信号通路。WNT信号可激活四种不同的通路:典型的WNT/β-CATENIN级联;非经典平面电池极性;WNT/Ca公司2+; 和蛋白激酶A途径(25,27). WNT分子与同源受体结合,该同源受体由七程跨膜Frizzled受体和位于细胞表面的相关单程低密度脂蛋白受体相关蛋白(LRP5/6)组成,在典型(WNT/β-CATENIN)途径中,激活细胞内Disheveled蛋白并将AXIN蛋白募集到细胞膜。因此,AXIN–APC–GSK3β–CK1α破坏复合物变得不活跃,β-CATENIN的细胞溶质水平增加,从而导致β-CATEN移位到细胞核。βCATENIN进入细胞核后,与T细胞因子(TCF)/淋巴增强因子(LEF)转录因子家族成员以及B细胞CLL/淋巴瘤9(BCL9)和皮戈普斯取代分裂/HDAC1阻遏蛋白的GROUCHO/转导蛋白样增强子,并通过招募染色质重塑子(如CBP/p300、MLL1/MLL2和BRG1)刺激靶基因表达(20,21,26).
WNT抑制剂基因的遗传缺失、点突变和表观遗传沉默导致的畸变导致WNT/β-CATENIN增殖信号的不受控激活和持续(20,22,25). WNT/β-CATENIN信号通路的失调是多种肿瘤的标志,包括淋巴瘤、白血病、结直肠癌和肝细胞癌(28). 虽然已知WNT/β-CATENIN拮抗剂启动子区的DNA甲基化导致其转录沉默,并与β-CATENIN/TCF/LEF靶基因的激活和肿瘤的发生有关,但对PRMT5在这一过程中的作用知之甚少。使用患者衍生的NHL细胞系,我们发现PRMT5通过两种主要拮抗剂的直接转录抑制促进WNT/β-CATENIN信号传导,AXIN2型和WIF1系列和AKT/GSK3β信号的间接激活。用短发夹RNA(shRNA)介导的敲低或PRMT5特异性抑制剂化合物5(CMP-5)抑制PRMT5,导致AXIN2型和WIF1系列磷酸化AKT(Thr-450和Ser-473)的再表达、失活和GSK3β的再激活。这些分子变化的最终结果是WNT/β-CATENIN靶基因的表达减少循环蛋白D1,c-MYC公司、和苏维文诱导淋巴瘤细胞死亡。招募研究表明,PRMT5抑制导致H3(Me)丢失2)R8和H4(Me2)基因启动子区的R3甲基化标记AXIN2型和WIF1系列此外循环蛋白D1,c-MYC公司、和苏维文启动子,包括联合激活剂CBP、p300和MLL1的招募减少,以及它们诱导的表观遗传标记H3K9Ac、H3K14Ac和H3(Me三)K4,伴随着辅阻遏物HDAC2和LSD1的结合增强。这些结果在NHL患者样本和小鼠原发性淋巴瘤细胞中也得到了证实。因此,我们的工作将PRMT5确定为来自多种淋巴瘤组织学亚型的细胞中AKT/GSK3β和WNT/β-CATENIN信号通路的新调节器,并进一步验证了靶向PRMT5在侵袭性淋巴瘤中的重要性。
结果
PRMT5调节WNT/β-CATENIN靶基因的表达
我们之前已经证明,正常B细胞中PRMT5蛋白水平较低,并且由于特定miRNAs表达减少,其水平升高(10,11). 我们之前也报道过,PRMT5通过灭活视网膜母细胞瘤抑癌蛋白RB1和RBL2以及上调PRC2来促进淋巴瘤细胞的生长和存活(12). 我们之前的工作还表明,尽管RB1抑制是由于CYCLIN D1–CDK4/CDK6的过度磷酸化所致,但RBL2失活是PRMT5介导的表观遗传沉默的直接结果。此外,对蛋白水平的评估表明,PRMT5表达增加与CYCLIN D1(一种已知的WNT/β-CATENIN靶基因)水平升高之间存在强烈的相关性(12). 根据这些结果和发现,PRMT5对c-MYC驱动的肿瘤发生至关重要(29),我们假设PRMT5可能通过促进WNT/β-CATENIN信号传导来影响淋巴瘤细胞的生长和增殖。
为了验证PRMT5是否参与调节WNT/β-CATENIN信号传导,我们测量了WNT/α-CATENIN下游靶基因的mRNA和蛋白水平,循环蛋白D1,c-MYC公司、和苏维文(图1). 使用两种不同的患者衍生细胞系评估所选三种不同淋巴瘤细胞类型中PRMT5和WNT/β-CATENIN靶基因之间的关系,包括MCL(Mino和JeKo)、GCB-DLBCL(Pfeiffer和Toledo)和ABC-DLBCL。我们发现,尽管在正常和转化的B细胞中,β-CATENI mRNA和蛋白表达没有显著变化,但CYCLIN D1增加(1.7–2.5倍,第页< 10−3),c-MYC(2–3.3倍,第页< 10−3)和SURVIVIN(3-4倍,第页< 10−3)与对照正常静息或活化CD19+B细胞相比,转化的B细胞中的mRNA水平和蛋白表达(图1,一个和B类). 与我们之前的发现一致,PRMT5蛋白水平在所有三种淋巴瘤细胞类型中也升高。有趣的是,PRMT5蛋白表达与WNT/β-CATENIN靶基因水平增强之间的正相关与核和转录活性磷酸化-β-CATEN(Ser-675)数量的增加平行(图1C类)表明PRMT5通过激活WNT/β-CATENIN-信号通路促进淋巴瘤细胞生长和存活。
图1。
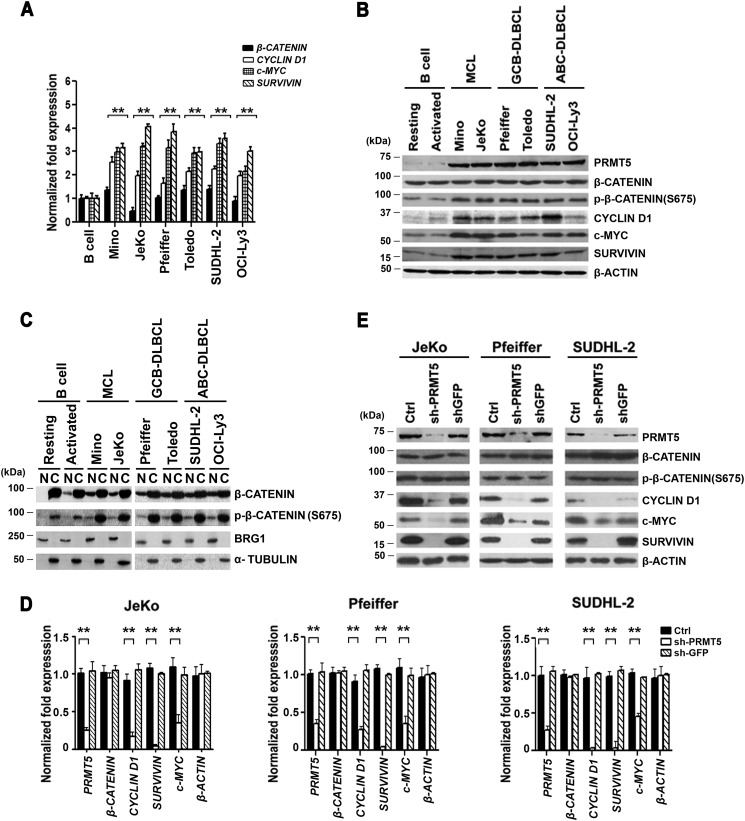
PRMT5调节患者衍生淋巴瘤细胞系中的WNT/β-CATENIN信号。
A、,的级别PRMT5项目, β-卡特宁,循环蛋白D1,c-MYC公司、和苏维文使用基因特异性引物和探针集,通过实时RT-PCR在正常B细胞(静止或激活)、GCB前MCL(Mino和JeKo)、GCB-DLBCL(Pfeiffer和Toledo)和GCB后ABC-DLBCL细胞(SUDHL-2和OCI-Ly3)中检测mRNA。这些值表示三个生物复制品的平均值,每个生物复制品有三个技术复制品,并以平均值±S.D.的形式报告。18秒rRNA被用作内部对照。B中,使用所示抗体通过蛋白质印迹分析来自正常或转化B细胞的RIPA提取物(20μg)。β-肌动蛋白用作负荷控制,用于B类和图2D类因为这些数据来自同一个实验。该实验重复了两次,并显示了具有代表性的斑点。C、,约25μg核(N个)或细胞溶质(C类)从正常休眠或激活的B细胞以及指示的淋巴瘤细胞系中制备提取物,并使用指示的抗体检测蛋白质。BRG1和α-管蛋白都可以作为核和细胞溶质分馏的对照。D、,的级别PRMT5项目, β-卡特宁,循环蛋白D1,c-MYC公司、和苏维文mRNA通过实时RT-PCR检测,使用从未感染的任何一种中分离的总RNA(Ctrl键)JeKo、Pfeiffer和SUDHL-2细胞,或感染shGFP或shPRMT5慢病毒的淋巴瘤细胞。使用特异性引物和探针组来检测每个指示的基因。本实验使用三个生物复制品和三个技术复制品进行,如一个.**表示第页值<10−3.E、,从对照感染和感染(shGFP或shPRMT5)淋巴瘤细胞系中制备RIPA提取物(20μg),并使用指示的抗体进行免疫印迹分析。β-肌动蛋白用作负荷控制。
为了研究PRMT5对WNT/β-CATENIN靶基因表达的影响程度,我们通过shRNA介导的敲除抑制其在JeKo、Pfeiffer和SUDHL-2细胞中的表达,并监测CYLIN D1、c-MYC和SURVIVIN的mRNA和蛋白表达。我们之前已经证明,使用两种不同的shRNAs(shPRMT5-1和shPRMT5-2)敲除PRMT5,可以实现PRMT5 mRNA和蛋白质水平的类似降低(12). 因此,我们选择在本研究中仅使用shPRMT5-1。mRNA和蛋白质分析均表明,PRMT5表达降低导致细胞周期蛋白D1的抑制(3-5倍,第页< 10−3),c-MYC(2.3–3倍,第页< 10−3)和SURVIVIN(26-50倍,第页< 10−4)所有三种淋巴瘤细胞类型的mRNA水平和蛋白表达与对照未感染淋巴瘤细胞或感染慢病毒的淋巴瘤细胞(表达对照shGFP)的比较(图1,D类和E类). 为了进一步证实这些结果,我们使用了PRMT5特异性抑制剂化合物5(CMP-5),最近对其进行了表征(13),用于治疗JeKo、Pfeiffer和SUDHL-2细胞系(图S1,一个和B类). 与PRMT5敲除实验一致,在所有三种淋巴瘤细胞类型中,CYCLIN D1、c-MYC和SURVIVIN mRNA和蛋白表达均明显降低,但在接受DMSO或对照CMP-6治疗的对照淋巴瘤细胞中没有降低。此外,当我们分析经CMP-5处理的JeKo、Pfeiffer和SUDHL-2细胞的增殖和存活时,细胞生长在第2天受到明显抑制,在第6天达到最大值,并最终导致所有三种淋巴瘤细胞类型的细胞死亡加剧,如膜联蛋白V阳性细胞数量的增加所证明的那样(图S1,C类和D类). 总的来说,这些结果表明,PRMT5控制前生存因子CYCLIN D1、c-MYC和SURVIVIN的表达,并且其抑制导致其抑制和诱导淋巴瘤细胞死亡。
PRMT5表观遗传抑制WNT/β-CATENIN拮抗剂、AXIN2和WIF1
鉴于WNT/β-CATENIN信号在细胞生长和增殖调节中的重要性,我们想确定PRMT5是否直接参与控制通路拮抗剂的表达。为了评估PRMT5的全基因组分布,使用免疫抗H3(Me2)来自Pfeiffer细胞的免疫沉淀交联染色质的R8抗体。在确定的共同靶基因中(表S1),我们发现H3R8甲基化在AXIN1型,AXIN2、,和WIF1系列与对照输入DNA相比(图2一个). 为了确认ChIP-Seq结果,对使用免疫前或免疫性抗PRMT5、抗H3(Me2)R8和抗H4(Me2)R3级(图2B类和图S2,一个和B类). 与ChIP-Seq结果一致,有2-3倍(第页< 10−4)PRMT5募集增加,表观遗传标记在AXIN1型,AXIN2型、和WIF1系列此外,对所有三种WNT/β-CATENIN拮抗剂的启动子区扩增区的检查显示,p53、PAX5、TFII-I和雌激素受体-α(ER-α)转录因子存在保守的DNA结合位点(表S2).
图2。
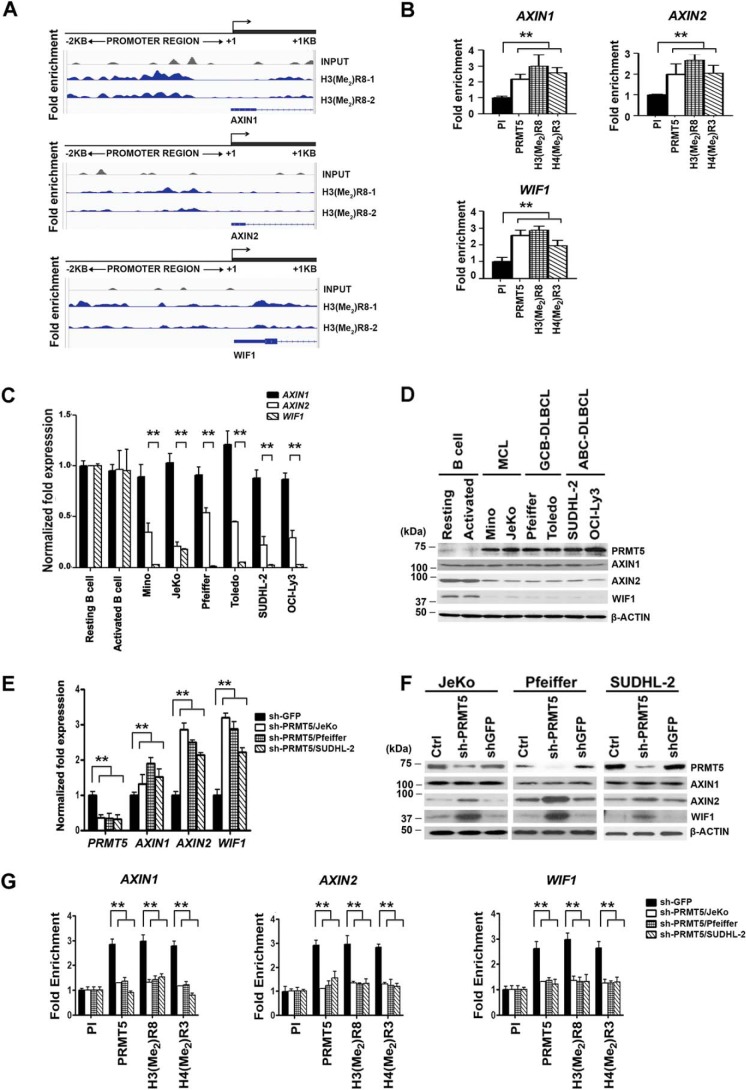
PRMT5表观遗传调节AXIN2型和WIF1系列,并且它的抑制激活了两个靶基因。
A、,使用抗H3(Me)抗体免疫沉淀Pfeiffer细胞的交联染色质2)R8抗体,并由ChIP-Seq按照“实验程序”进行分析。H3(Me)的富集2)R8开启AXIN1型,AXIN2型、和WIF1系列使用两个生物复制品(H3(Me2)R8-1和H3(Me2)R8-2)。H3(Me的峰值检测2)报告R8富集蓝色与控制输入相比,后者在灰色。B中,使用预免疫对Pfeiffer细胞的交联染色质进行ChIP分析(圆周率)、抗PRMT5、抗H3(Me2)R8或抗H4(Me2)R3抗体。ChIP实验使用两个生物复制品和三个技术复制品进行AXIN1型,AXIN2型、和WIF1系列通过实时PCR检测启动子序列。相对于PI样品计算每个抗体的折叠富集。每个中的数据图表表示平均值±S.D。C、,的级别AXIN1型,AXIN2型、和WIF1系列使用基因特异性引物和探针集,通过实时RT-PCR在正常B细胞(静止和激活)和指示的转化B细胞中测量mRNA。本实验采用三个生物重复和三个技术重复进行18秒rRNA作为对照。所示数据代表平均值±S.D。D、,使用所示抗体通过Western blotting分析正常(静止和激活)和转化B细胞的RIPA提取物(20μg)。抗β-肌动蛋白用于显示等负荷。β-肌动蛋白负荷控制B类和D类是相同的,因为这些数据来自同一个实验。E、,的级别PRMT5项目,AXIN1型,AXIN2型、和WIF1系列用两个对照组的总RNA测量mRNA(Ctrl键)未感染的JeKo、Pfeiffer和SUDHL-2细胞或感染shGFP或shPRMT5慢病毒的淋巴瘤细胞C类.**表示第页值<10−3.F、,使用从感染后72 h的对照未感染和感染(shGFP或shPRMT5)JeKo、Pfeiffer和SUDHL-2淋巴瘤细胞系制备的RIPA提取物(20μg),通过Western blotting分析AXIN1、AXIN2和WIF1蛋白水平。抗β-肌动蛋白用于显示等负荷。G中,使用来自对照shGFP或shPRMT5感染淋巴瘤细胞系的交联染色质进行ChIP分析。使用指示的抗体测定PRMT5及其诱导的表观遗传标记的富集,并使用PI抗体作为对照。使用三个生物复制品重复该实验,每个生物复制品有三个技术复制品,数据如B类.
根据这些结果,我们继续评估WNT/β-CATENIN拮抗剂在对照静止或激活的B细胞和转化的B细胞系中的mRNA和蛋白表达(图2,C类和D类). AXIN2和WIF1 mRNA和蛋白质均表现出2–20倍(第页< 10−3)与对照B细胞相比,淋巴瘤细胞的表达谱降低,而AXIN1 mRNA和蛋白水平在正常和转化的B细胞中不受影响。为了验证PRMT5抑制是否改变AXIN2和WIF1的表达,我们敲除了其在JeKo、Pfeiffer和SUDHL-2细胞中的表达,并监测其mRNA和蛋白水平(图2,E类和F类). 两者都有AXIN2型和WIF1系列显示2-3倍(第页< 10−3)mRNA水平的去表达,与未感染或shGFP感染的对照淋巴瘤细胞相比,其蛋白表达增加。与对照组β-肌动蛋白一样,AXIN1 mRNA和蛋白水平没有任何显著变化。同样,当我们使用CMP-5抑制PRMT5时,有1.8–3倍(第页< 10−3)解除两者的压力AXIN2型和WIF1系列mRNA及其蛋白表达适度增加(图S3,一个和B类).
发现PRMT5在所有三个WNT/β-CATENIN拮抗剂基因的启动子区域富集,我们试图监测其在对照和PRMT5敲除以及CMP-5治疗的淋巴瘤细胞系中的募集(图2G公司和图S3C类). 与对照shGFP感染的淋巴瘤细胞和DMSO治疗的淋巴瘤细胞相比,PRMT5敲除细胞和CMP-5治疗淋巴瘤细胞系中PRMT5的招募和表观遗传标记的富集均被消除。综上所述,这些结果表明PRMT5表观遗传学抑制AXIN2型和WIF1系列促进WNT/β-CATENIN信号传导和靶基因表达。
AXIN2和/或WIF1再表达导致AKT/GSK3β磷酸化和CYCLIN D1、c-MYC和SURVIVIN表达降低
为了评估AXIN2和WIF1在WNT/β-CATENIN信号转导中的作用,我们体外恢复了它们在Pfeiffer细胞中的单独或联合表达,并监测了它们的表达循环蛋白D1,c-MYC公司、和苏维文(图3). 收集总RNA和全细胞提取物,并检测其mRNA和蛋白表达。重新表达AXIN2型或WIF1系列引发了循环蛋白D1(2.2-2.9倍,第页< 10−4),c-MYC公司(3–3.8倍,第页< 10−4)、和苏维文(3–7.7倍,第页< 10−3)mRNA水平(图3,一个和B类)以及蛋白质表达的减少(图3C类). 值得注意的是,两种WNT/β-CATENIN拮抗剂的共同表达导致了更显著的下降(3.8–8.3倍,第页< 10−4)WNT/β-CATENIN靶基因mRNA水平,伴随着其蛋白表达的更显著下降。
图3。
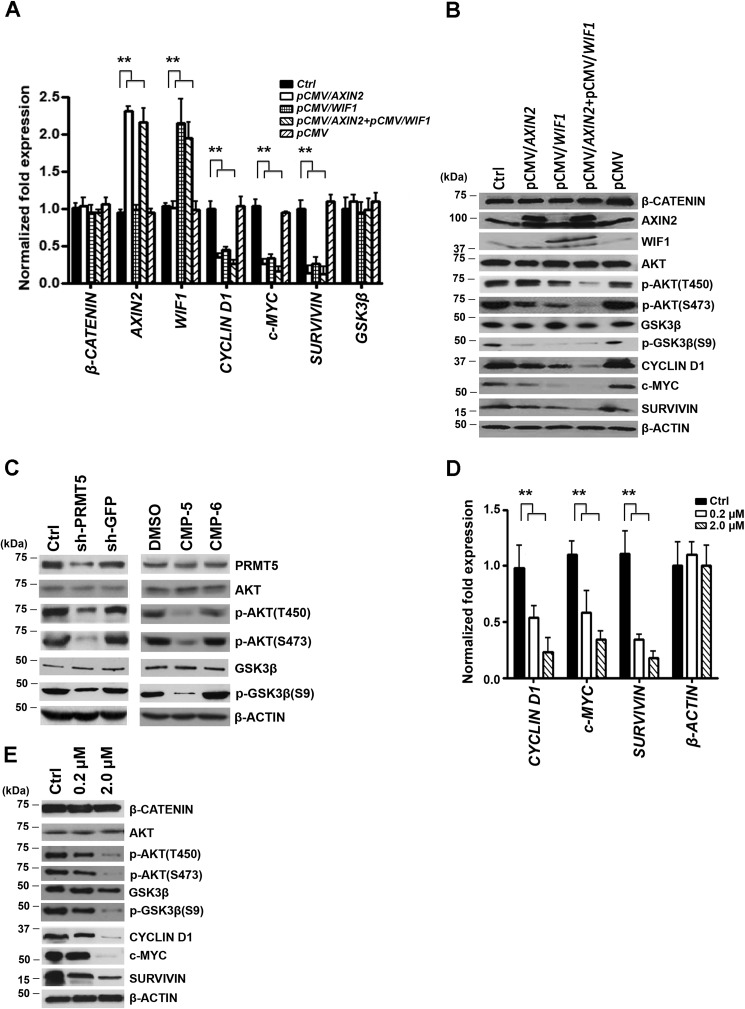
AXIN2和/或WIF1的再表达通过AKT失活抑制WNT/β-CATENIN靶基因的表达。
A、,用任一对照转染Pfeiffer细胞(Ctrl键)载体(pCMV-entry)或pCMV-entry/AXIN2和/或pCMV entry/WIF1 72小时,以及AXIN2型,WIF1系列,循环蛋白D1,c-MYC公司、和苏维文通过实时RT-PCR进行分析。本实验使用三个生物重复,每个重复三个技术重复,以及β-肌动蛋白用作对照。所示数据代表平均值±S.D.,**表示第页值<10−3.B中,用所示抗体通过免疫印迹分析从对照未感染或转染Pfeiffer细胞制备的RIPA提取物(20μg)。检测到β-肌动蛋白显示等负荷。C、,使用RIPA提取物(20μg)通过免疫印迹法测定活性磷酸化AKT(Thr-450或Ser-473)和非活性AKT以及活性GSK3β或非活性磷酸化GSK3?(Ser-9)的水平由用表达对照shGFP或shPRMT5的慢病毒感染的Pfeiffer细胞和用对照DMSO或CMP-5处理的Pfeiffer细胞制备。未感染和CMP-6处理的Pfeiffer细胞均被用作内部对照。在感染后72小时或治疗后48小时制备RIPA细胞提取物。D、,用控制二甲基亚砜或增加AKT抑制剂IV浓度(0.2和2μ米)和mRNA水平循环蛋白D1,c-MYC公司、和苏维文使用基因特异性引物和探针集通过实时RT-PCR进行评估,如A.E、,Pfeiffer细胞按照D类和RIPA提取物(20μg)使用所示抗体进行Western blotting分析。检测到β-肌动蛋白显示等负荷。
众所周知,标准WNT信号调节转录活性β-CATENIN的量(30). 众所周知,在许多肿瘤细胞类型中被激活的AKT(蛋白激酶B)通过糖原合成酶激酶3β(GSK3β)的磷酸化和失活与WNT/β-CATENIN信号相互作用,从而促进转录活性磷酸化β-CATEN(Ser-675)的核移位(31,32). 根据这些研究,我们评估了对照和转染Pfeiffer细胞中AKT/GSK3β的表达和磷酸化(图3B类). 重新表达AXIN2型或WIF1系列导致磷酸化AKT(Thr-450和/或Ser-473)水平略有下降;然而,在两种WNT拮抗剂存在的情况下,通过Thr-450和Ser-473的去磷酸化测定,活性AKT显著下降(33). 类似地,重新表达AXIN2型和或WIF1系列导致磷酸化GSK3β(Ser-9)水平降低,这直接反映了AKT的失活。因为我们的研究结果表明PRMT5抑制作用减弱AXIN2型和WIF1系列,我们想确定PRMT5敲除或用CMP-5抑制PRMT5对AKT/GSK3β磷酸化是否有相同的影响(图3C类). 事实上,这两种治疗都降低了Thr-450和Ser-473的AKT磷酸化以及Ser-9的GSK3β磷酸化,证实了AXIN2型和WIF1系列重新表达结果。
为了进一步评估AKT/GSK3β信号传导对WNT/β-CATENIN靶基因表达的调节作用,我们使用增加AKT抑制剂IV浓度(0.2和2μ米)为了治疗Pfeiffer细胞,我们监测了CYCLIN D1、c-MYC和SURVIVIN mRNA和蛋白的表达(图3,D类和E类). 尽管AKT抑制剂IV浓度较低(0.2μ米)减少了2-3倍(第页< 10−3)在所有三个WNT/β-CATENIN靶基因的mRNA表达中,其蛋白表达没有显著下降。然而,当2μ米AKT抑制剂IV的使用量减少了4倍(第页< 10−4)mRNA水平和CYCLIN D1、c-MYC和SURVIVIN蛋白表达显著下降。同样,在2μ米这些发现表明AXIN2和WIF1协同下调AKT抑制剂IV的表达循环蛋白D1,c-MYC公司、和苏维文通过AKT/GSK3β和WNT/β-CATENIN信号的失活。
AXIN2和WIF1去表达动力学与AKT/GSK3β去磷酸化和WNT/β-CATENIN靶基因表达沉默一致
发现PRMT5抑制可恢复两者的表达AXIN2型和WIF1系列并导致AKT/GSK3β去磷酸化,我们试图确定这些事件发生的时间。在敲低PRMT5表达或抑制其活性后进行时间过程分析(图4和图S4). 当通过短发夹介导的敲除降低PRMT5水平时,AXIN2型和WIF1系列感染后36小时,mRNA水平增加(1.5倍,第页< 10−3)达到2–2.2倍(第页< 10−3)感染后72小时降压(图4一个). mRNA水平的变化与AXIN2和WIF1蛋白水平的增加相关(图4C类). 与这些结果一致循环蛋白D1,c-MYC公司、和苏维文mRNA水平,感染后36–48小时开始,感染后72小时达到最高水平(图4B类). 然而,直到感染后48–60小时,WNT/β-CATENIN靶基因蛋白表达才明显下降。此外,对AKT/GSK3β磷酸化的分析表明AXIN2型和WIF1系列去加压和AKT/GSK3β去磷酸化(图4C类).
图4。
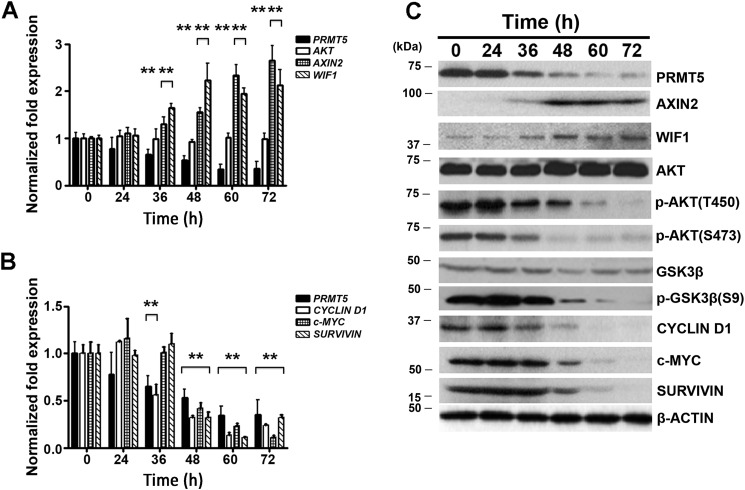
PRMT5敲除通过AXIN2和WIF1的重新表达以及AKT信号的失活触发WNT/β-CATENIN靶基因抑制。Pfeiffer细胞感染表达shPRMT5的慢病毒PRMT5、AKT、AXIN2、,和WIF1系列(一个)、和PRMT5,循环D1,c-MYC公司、和苏维文(B类)通过实时RT-PCR在不同时间点使用18秒rRNA作为内部控制。为了进行比较,在感染前0 h测定mRNA水平。数值表示三个生物复制品的平均值,每个生物复制品有三个技术复制品,报告为平均值±S.D。C、,从对照未感染(0小时)和感染(shPRMT5)的Pfeiffer细胞系中制备RIPA提取物(20μg),并使用指示的抗体通过免疫印迹进行分析。检测到β-肌动蛋白显示等负荷。
当用CMP-5抑制PRMT5时,也观察到类似的结果;然而,再表达的动力学AXIN2型(1.4–2.5倍,第页< 10−4)和WIF1系列(1.5–2.9倍,第页< 10−4)与PRMT5表达被抑制的细胞相比,mRNA更快(感染后1-3小时)(图S4一个). 此外,AXIN2和WIF1蛋白水平同时增加,这在WIF1治疗后1小时更为明显(图S4C类). 当我们检测CYCLIN D1、c-MYC和SURVIVIN mRNA和蛋白的表达时,我们发现它们在治疗后6-12小时开始减少,进一步证实了AXIN2型和WIF1系列再表达先于抑制WNT/β-CATENIN靶基因表达(图S4,B类和C类). 我们的结果还表明,在恢复表达AKT/GSK3β后立即(治疗后3小时)发生去磷酸化AXIN2型和WIF1系列表明这两种拮抗剂协同作用,使AKT/GSK3β和WNT/β-CATENIN信号失活。
WNT/β-CATENIN靶基因的启动子在抑制PRMT5后经历特异的赖氨酸脱乙酰和脱甲基
转录调控循环蛋白D1,c-MYC、,和苏维文已知通过β-CATENIN与其启动子的结合上调(34). 事实上,我们已经发现PRMT5基因敲除或其甲基转移酶活性的抑制导致WNT/β-CATENIN靶基因表达的显著降低,我们开始研究其启动子区域发生的动态变化(图5和图6). 首先,我们监测了β-CATENIN在细胞启动子区的募集循环蛋白D1,c-MYC公司、和苏维文在正常和转化的B细胞中(图5). β-CATENIN与所有三个WNT/β-CATEN靶基因的启动子近端区域的结合富集了3倍(第页< 10−4)淋巴瘤细胞与对照前免疫抗体的比较(第页< 10−3)与对照正常B细胞相比(图5一个). 然而,当PRMT5被击倒时(图5B类)或被抑制(图5C类),β-CATENIN在启动子区的募集没有显著变化循环蛋白D1,c-MYC公司、和苏维文.
图5。
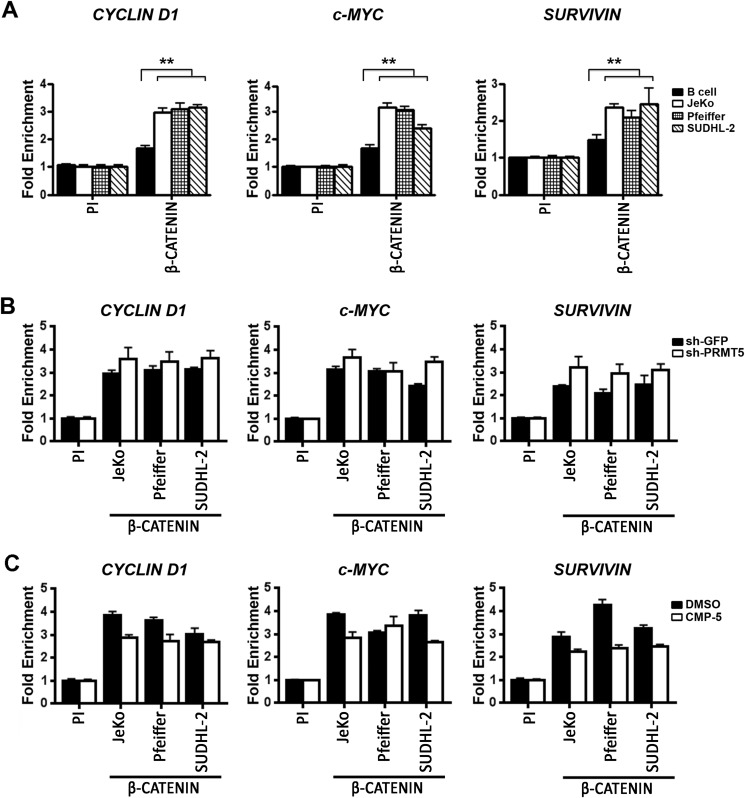
β-CATENIN募集到循环蛋白D1,c-MYC公司、和苏维文在淋巴瘤细胞中增加,并且不受PRMT5敲除的影响。
A、,ChIP测定使用来自正常或指示的转化B细胞的交联染色质进行,使用PI或免疫抗β-CATENIN抗体。本实验使用三个生物复制品进行,每个生物复制品有三个技术复制品循环蛋白D1,c-MYC公司、和苏维文实时PCR检测启动子序列。相对于PI样品计算折叠富集度,每个样品中的数据图表表示为平均值±S.D。B中,使用PI或免疫抗β-CATENIN抗体免疫沉淀来自感染慢病毒的指示淋巴瘤细胞的交联染色质,慢病毒表达控制性shGFP或shPRMT5。每个中的值图表使用两个生物复制品生成,每个复制品有三个技术复制品,如A.C中,用二甲基亚砜或CMP-5处理JeKo、Pfeiffer和SUDHL-2细胞,用PI或免疫抗β-CATENIN抗体免疫沉淀交联染色质。循环蛋白D1,c-MYC公司、和苏维文启动子序列检测如B类.
图6。
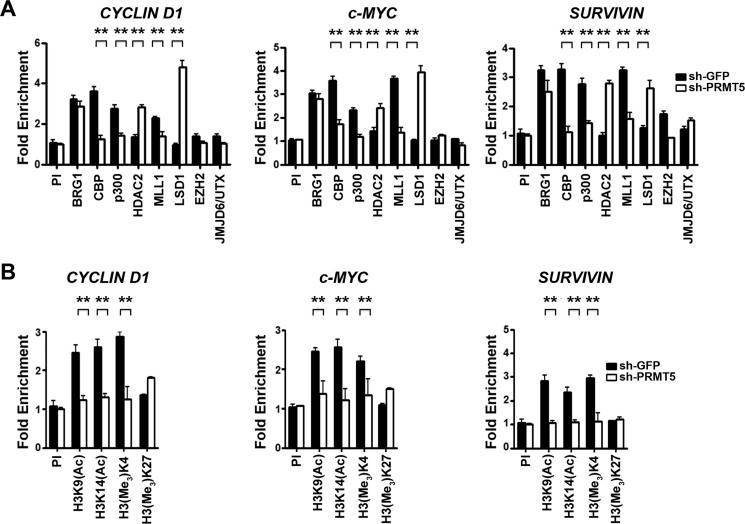
PRMT5基因敲除改变WNT/β-CATENIN靶基因的共激活物和共阻遏物的募集。
一个和B中,使用所示抗体对感染慢病毒的Pfeiffer细胞的交联染色质进行免疫沉淀,慢病毒表达对照shGFP或shPRMT5,PI抗体用作对照。使用两个生物复制品和三个技术复制品进行ChIP分析。计算每个抗体相对于PI样品的折叠富集度,以及每个抗体中的数据图表表示为平均值±S.D。
因为β-CATENIN的结合通过与多种转录共激活物的特异性结合诱导靶基因的转录(30,34),我们测量了在循环蛋白D1,c-MYC公司、和苏维文在对照组和PRMT5击倒淋巴瘤细胞中(图6和图S5). 所检测的蛋白质包括BRG1(hSWI/SNF染色质重塑复合物的催化亚单位)、CBP、p300组蛋白乙酰转移酶(乙酰化组蛋白H3赖氨酸9(H3K9)和H3K14)以及MLL1(专门甲基化H3K4并与转录激活相关)。ChIP分析还用于检查组蛋白H3和H4翻译后修饰以及共抑制蛋白的招募,包括HDAC2(拮抗CBP和p300)和LSD1(去甲基化H3K4并与转录抑制相关)。还检测了可通过H3K27甲基化诱导转录沉默的PRC2催化亚单位EZH2及其拮抗性去甲基化酶JMJD6/UTX的招募情况。
用免疫前或特异性免疫抗体对感染慢病毒的Pfeiffer细胞中表达控制性shGFP或shPRMT5的交叉染色质进行免疫沉淀,并通过实时PCR评估WNT/β-CATENIN靶基因启动子区的蛋白结合和组蛋白修饰(图6). 尽管BRG1、EZH2和JMJD6/UTX的募集没有显著变化,但CBP与循环1,c-MYC公司、和苏维文减少了2-3倍(第页< 10−3)在PRMT5敲除细胞中(图6一个). 同样,p300和MLL1的募集也减少了2倍(第页< 10−4)和1.7–2.7倍(第页< 10−3)分别在PRMT5敲除细胞中。然而,HDAC2和LSD1共同抑制蛋白与WNT/β-CATENIN靶基因启动子区的关联性增加了1.7-2.8倍(第页< 10−3)和2-5倍(第页< 10−3)分别是。当我们检测组蛋白H3和H4乙酰化和甲基化标记时,我们发现协同激活物募集的丢失与与基因激活相关的表观遗传标记的减少之间存在正相关(图6B类). 与转录激活相关的所有三个组蛋白标记都减少了1.8–2.8倍(第页< 10−3)对于H3K9(Ac),2–2.2倍(第页< 10−3)对于H3K14(Ac)和1.6–2.6倍(第页< 10−4)对于H3(Me三)WNT/β-CATENIN靶基因启动子区的K4。在H3K27病例中未观察到H3K4甲基化的降低,这突出了在循环蛋白D1,c-MYC公司、和苏维文发起人。当PRMT5在JeKo和SUDHL-2细胞中被敲除时,也观察到类似的结果(图S5,一个和D类),表明一种共同的机制被用来抑制循环蛋白D1,c-MYC公司、和苏维文在所有三种淋巴瘤细胞系中。
PRMT5介导的AXIN2和WIF1表观遗传沉默与NHL临床样本中AKT/GSK3β和WNT/β-CATENIN信号的组成性激活相关
我们使用三种不同类型的患者衍生淋巴瘤细胞系的研究结果清楚地表明,PRMT5水平升高会触发WNT/β-CATENIN拮抗剂的抑制,这反过来又有助于AKT/GSK3β和WNT/?CATENIN增殖信号的组成性激活。基于这些结果,我们想确定这些分子变化是否也发生在原代NHL肿瘤细胞中(图7). 从对照正常CD19+B淋巴细胞或三个NHL临床样本中制备总mRNA和蛋白提取物,并表达PRMT5项目以及AXIN2型和WIF1系列已评估。尽管PRMT5项目mRNA水平要么保持不变,要么略微增加1.9–2.5倍(第页< 10−2)与正常对照B细胞相比,在三种原发性肿瘤中的两种中,PRMT5蛋白在所有三种原发病中的表达都明显较高(图7,一个和C类). 相比之下AXIN2型和WIF1系列mRNA被抑制3.3–6.7倍(第页< 10−2到10−3)和8.3-20倍(第页< 10−2)分别在原代肿瘤细胞中。与这些结果一致,AXIN2和WIF1蛋白在原代肿瘤细胞中的表达也被高度抑制。
图7。
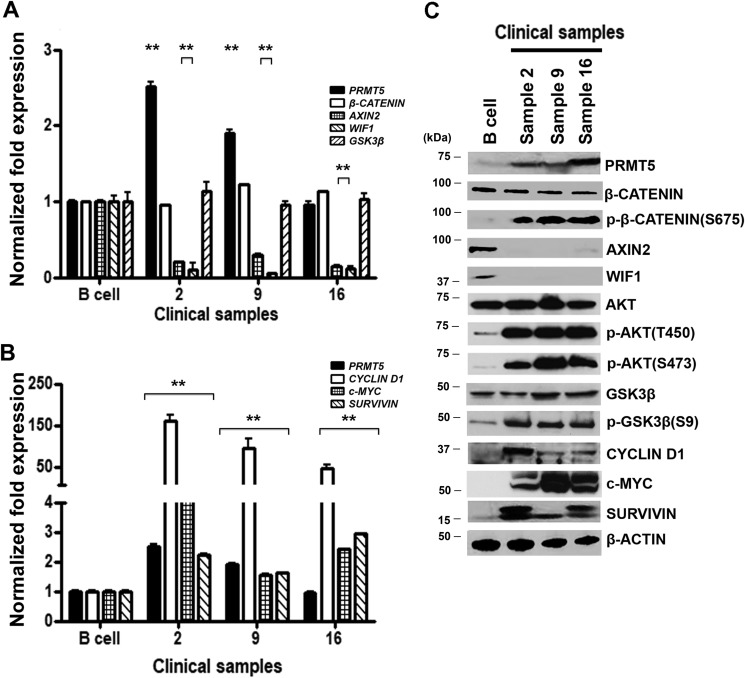
PRMT5过度表达与NHL临床样本中WNT/β-CATENIN和AKT/GSK3β信号增强相关。的级别PRMT5项目, β-卡特宁,AXIN2型,WIF1系列,葛兰素史克3β (一个)和循环蛋白D1,c-MYC公司、和苏维文(B类)在正常B细胞和NHL患者样本中检测mRNA(2, 9,和16)通过使用基因特异性引物和探针组的实时RT-PCR。本实验使用三个生物重复,每个重复三个技术重复,数值代表平均值±S.D。18秒rRNA被用作内部对照。C、,从正常B细胞和NHL患者样本中制备的RIPA提取物(40μg)用所示抗体进行免疫印迹分析,并检测β-肌动蛋白以显示等负荷。
由于AKT/GSK3β和WNT/β-CATENIN信号在患者衍生淋巴瘤细胞系中都是活跃的,我们研究了AKT、GSK3?和?CATENIN的磷酸化状态(图7C类). 与正常B细胞相比,原发性肿瘤细胞中通过Thr-450和Ser-473磷酸化测定的AKT活化显著增加,并与GSK3β(Ser-9)磷酸化增强和失活相关。接下来,我们检查了转录活性磷酸化-β-CATENIN(Ser-675)以及WNT/β-CATENIN靶基因的水平。我们发现,所有三个临床样本中磷酸化-β-CATENIN(Ser-675)的水平均升高,表明WNT/β-CATENIN增殖信号被激活。我们还发现,除了循环蛋白D1mRNA水平极高,为47至162倍(第页< 10−3)与正常B细胞相比增加,c-MYC公司和苏维文mRNA水平是1.6–4.6倍(第页< 10−3)原发性肿瘤细胞分别高1.6–2.9倍(图7B类). 与正常B细胞相比,这三个临床样本中的细胞周期蛋白D1、c-MYC和SURVIVIN蛋白表达也随着mRNA水平的增加而升高。这些结果支持了我们对患者源性淋巴瘤细胞系的研究结果,并进一步证实了PRMT5表达增强、AKT/GSK3β和WNT/β-CATENIN信号传导组成增强与肿瘤细胞表达增强之间的正相关循环蛋白D1,c-MYC公司、和苏维文生存因素。
抑制PRMT5通过AKT/GSK3β和WNT/β-CATENIN增殖信号的失活诱导小鼠原发性淋巴瘤细胞死亡
我们已经证明,PRMT5通过抑制WNT拮抗剂的表达和增强WNT/β-CATENIN靶基因在三种不同类型的患者衍生淋巴瘤细胞系和临床样本中的表达,促进淋巴瘤细胞的生长和存活。根据这些结果,我们试图确定PRMT5使人类淋巴瘤细胞增殖的机制是否也用于来自Eμ-BRD2转基因小鼠的小鼠原发性淋巴瘤细胞(35). 对Eμ-BRD2原发肿瘤细胞的转录组分析表明,在这种小鼠淋巴瘤中诱导的基因表达程序与人类DLBCL相似。因此,我们检查了项目5,轴2,无线1和WNT/β-CATENIN靶基因,我们还评估了AKT、GSK3β和β-CATEN的磷酸化状态(图8). 与我们在患者衍生细胞系和临床样本中的发现一致,项目5mRNA增加了4.3倍(第页< 10−3)与正常B细胞相比,Eμ-BRD2原发肿瘤细胞中PRMT5蛋白水平也升高(图8,一个和D类,左侧面板). 同样,细胞周期蛋白D1(1.9倍,第页< 10−3),c-MYC公司(2.7倍,第页< 10−3)、和Survivin公司(1.8倍,第页< 10−3)与正常B细胞相比,小鼠原发性淋巴瘤细胞的mRNA水平增加,其蛋白水平也增加。当我们检测WNT/β-CATENIN拮抗剂的表达时,我们发现两者都轴2和无线1mRNA分别被抑制7.7倍和8.3倍。然而,在蛋白水平上,与正常B细胞相比,原发性淋巴瘤细胞中AXIN2受到更显著的抑制,而WIF1仅在Eμ-BRD2淋巴瘤细胞中没有显示出任何减少。当我们评估AKT、GSK3β和β-CATENIN的磷酸化时,我们发现虽然AKT(Thr-450)磷酸化仅适度增强,但AKT(Ser-473)和β-CATENIN(Ser-675)磷酸化在淋巴瘤细胞中均显著诱导。相反,正常和小鼠原代Eμ-BRD2肿瘤细胞中GSK3β(Ser-9)的磷酸化没有改变。
图8。
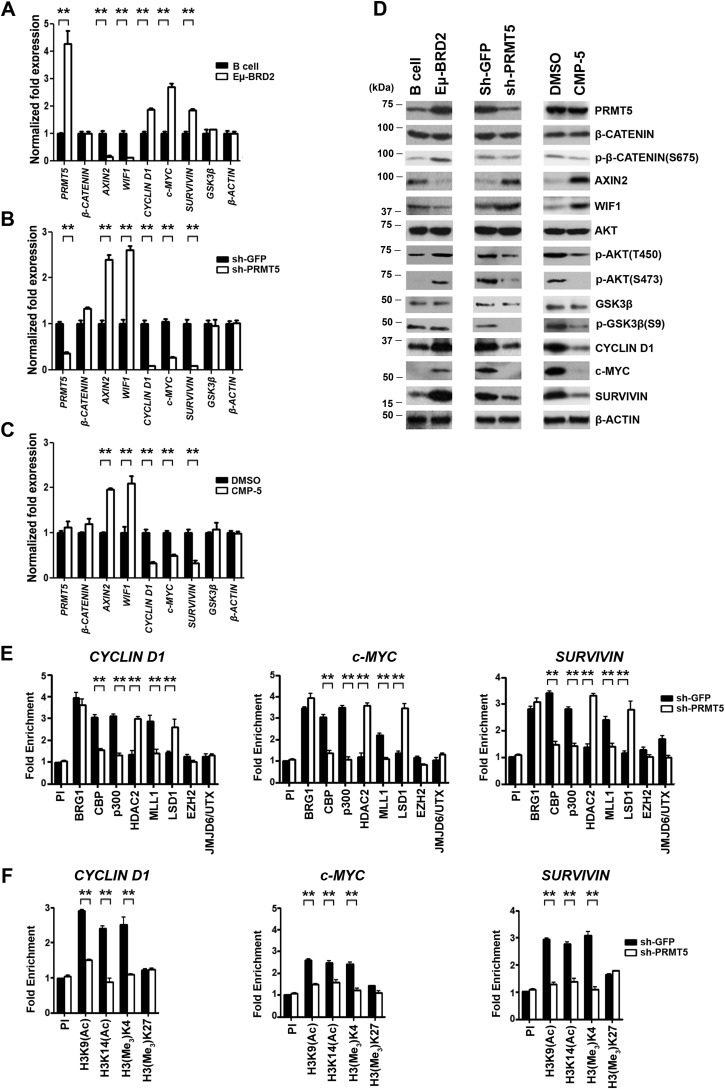
PRMT5敲除或抑制导致轴2和无线1小鼠原发性淋巴瘤细胞中WNT/β-CATENIN和AKT/GSK3β信号的去表达和失活。
A、,对正常小鼠B细胞和Eμ-BRD2细胞的总RNA进行实时RT-PCR,并使用基因特异性引物集和探针测量指示靶基因的稳态mRNA水平。B中,在感染表达控制shGFP–GFP或shPRMT5–GFP的慢病毒之前,通过添加重组人白细胞介素-4(15 ng/ml)和山羊抗人IgG+IgM(15μg/ml)激活Eμ-BRD2细胞。感染后72小时分离总RNA,并使用基因特异性引物和探针集通过实时RT-PCR测量指示基因的mRNA水平。C、,从用对照DMSO或CMP-5处理的Eμ-BRD2细胞中分离出总RNA,并在处理48小时后测量所示靶基因的mRNA水平,如出生日期:,从所示细胞中制备RIPA提取物(40μg),并使用指定抗体进行免疫印迹分析。β-肌动蛋白用作负荷控制。E类和F、,ChIP分析使用来自感染表达shGFP或shPRMT5慢病毒的活化Eμ-BRD2细胞的交联染色质进行。每个中的值图表由两个生物重复和三个技术重复生成,并绘制为平均值±S.D。
验证PRMT5是否控制细胞周期蛋白D1,c-Myc公司、和Survivin公司通过调节AKT/GSK3β和WNT/β-CATENIN信号,我们开始通过shPRMT5介导的敲除来抑制PRMT5(图8,B类和D类,中间面板)或CMP-5介导的抑制(图8,C类和D类,右侧面板).项目5击倒结果是2.4倍和2.6倍(第页< 10−4)解除对…的压制轴2和无线1mRNA水平,这与与对照shGFP相比观察到的蛋白表达增加一致(图8D类). 此外,WNT/β-CATENIN靶基因的转录减少了11.6倍(第页< 10−3)对于两者细胞周期蛋白D1和Survivin公司和3.8倍(第页< 10−3)对于c-Myc公司在蛋白质水平上,WNT/β-CATENIN靶基因表达下降,伴随着AKT(Ser-473)和GSK3β(Ser-9)的去磷酸化。当用PRMT5抑制剂治疗原发性淋巴瘤细胞时,WNT/β-CATENIN拮抗剂和靶基因的mRNA和蛋白表达也发生了类似的变化(图8,C类和D类,右侧面板). 两者都有轴2和无线1mRNA被释放2倍(第页< 10−3)蛋白表达的增加清楚地反映了这一点。此外,细胞周期蛋白D1和Survivin公司mRNA水平被抑制了3.1倍,而c-Myc公司与未经处理的细胞相比,经处理的原发性淋巴瘤细胞的mRNA减少了2倍。WNT/β-CATENIN靶基因mRNA水平的这些负变化也在蛋白质水平上检测到。当评估AKT和GSK3β的磷酸化时,磷酸化-AKT(Thr-450和Ser-473)和磷酸化-GSK3β(Ser-9)显著下降。
为了确定WNT/β-CATENIN靶基因表达的变化是否由同一组染色质重塑酶控制,我们进行了ChIP实验,以监测在启动子区的共激活酶和共抑制酶的结合细胞周期蛋白D1,c-Myc公司、和Survivin公司(图8,E类和F类). 招募CBP、p300和MLL1加入细胞周期蛋白D1启动子区域减少了2–2.4倍(第页< 10−3)在项目5敲除细胞,HDAC2和LSD1的结合增强了2.1倍和1.9倍(第页< 10−3)分别为(图8E类). 在c-Myc公司启动子,共激活物结合减少2.1倍(第页< 10−2)对于CBP,为3.2倍(第页< 10−3)p300和2.2倍(第页< 10−3)对于MLL1。此外,联合阻遏物招募受到3倍和2.5倍的刺激(第页< 10−3)分别用于HDAC2和LSD1。在Survivin公司启动子,其中共激活物的联合减少1.7–2.3倍(第页< 10−3)联合阻遏物的招募增加了2.3倍(第页< 10−3). 与这些分子变化一致,我们发现组蛋白H3乙酰化(H3K9Ac和H3K14Ac)和甲基化(H3Ce)降低三K4)在所有三个WNT/β-CATENIN靶基因的启动子区(图8F类). 在细胞周期蛋白D1启动子、H3K9/K14乙酰化和H3K4甲基化标记减少了2–2.8倍(第页< 10−3),而在c-Myc公司和Survivin公司启动子,乙酰化和甲基化标记的减少在1.6到2倍之间(第页< 10−3)以及2倍和3倍(第页< 10−3)分别是。表观遗传标记的这些波动是特定的,因为EZH2的补充和其诱导的H3(Me三)K27标记显示对照组shGFP和shPRMT5敲除细胞有任何变化。
讨论
PRMT5过度表达对癌细胞生长和生存的影响已在几种癌细胞类型中得到证实,其作用机制似乎多样,足以调节广泛的生长调节途径(16). 我们之前已经使用不同的患者衍生淋巴瘤细胞系探索了PRMT5在控制癌细胞生长中的作用,并且我们能够确定PRMT5作用于CYCLIN D1/CDK4/CDK6和RB/E2F通路的上游(12). 根据这些发现,PRMT5敲除或抑制导致循环蛋白D1下调和诱导细胞死亡,我们的ChIP-Seq和实时ChIP结果表明,PRMT5富集在WNT/β-CATENIN拮抗基因的启动子区域,我们研究了PRMT5抑制对WNT/α-CATENIN信号和下游靶基因表达的影响。
我们的结果表明,在三种不同的淋巴瘤细胞类型中,PRMT5的过度表达与CYCLIN D1、c-MYC和SURVIVIN蛋白水平的增加呈正相关(图1). NHL临床样本和小鼠原发性淋巴瘤细胞证实了这种相关性(图7和8). 当PRMT5被敲除或抑制时,所有三个WNT/β-CATENIN靶基因的mRNA和蛋白质水平均显著降低(图1和图S1). 此外,我们的ChIP-Seq结果显示,PRMT5与三个WNT/β-CATENIN拮抗基因的启动子区结合,AXIN1型,AXIN2型、和WIF1系列(图2和图S2). 实时ChIP分析验证了我们的发现,并促使我们检查患者衍生淋巴瘤细胞系、NHL临床样本和小鼠原发性淋巴瘤细胞中所有三个PRMT5靶基因的表达。我们发现,尽管所有三种WNT/β-CATENIN拮抗剂在mRNA水平上都受到抑制,但只有AXIN2型和WIF1系列在蛋白质水平上被抑制,表明还有其他机制参与调节AXIN1型表达式。评估PRMT5在转录抑制中的作用AXIN1型,AXIN2型、和WIF1系列,我们通过shRNA-介导的敲除抑制其甲基转移酶活性(图2,E–G公司)或CMP-5处理(图S3). 我们发现,在两种治疗方案下,WNT/β-CATENIN拮抗剂基因启动子区的PRMT5募集均被取消。因此,AXIN2型和WIF1系列在mRNA和蛋白质水平上被去压;然而,AXIN1型mRNA水平仅略有增加(<2倍),蛋白水平无变化,为其调控中存在其他机制提供了更多证据。
事实上,以前的工作表明AXIN1型通过启动子DNA超甲基化在肺癌和结直肠癌细胞中下调,增加了DNA甲基化参与调节的可能性AXIN1型基因表达(36,37). 另一个值得一提的有趣点是,最近一项关于结直肠癌的研究认为AXIN1型和AXIN2型β-CATENIN破坏复合物中(38). 本研究的结果表明,AXIN1不影响破坏复合物诱导的β-CATENIN降解,而AXIN2缺失会损害β-CATEN破坏复合物的功能,这意味着AXIN2在调节结直肠癌中WNT/β-CATENIN信号传导中起着更重要的作用。我们的研究结果表明,AXIN2在PRMT5抑制后变得去压,这表明这可能会增强β-CATENIN破坏复合物的功能,并导致β-CATENIN水平降低。然而,当患者源性细胞系中PRMT5被敲除或抑制时,我们的结果并未显示β-CATENI或磷酸化β-CATEN(Ser-675)稳态水平的任何变化(图1E类)或小鼠原发性淋巴瘤细胞(图8D类). 当AXIN2在淋巴瘤细胞中重新表达时,总β-CATENIN的量也没有变化(图3B类). 同样,实时ChIP分析测定的染色质结合β-CATENIN水平也没有下降(图5)表明淋巴瘤细胞中WNT/β-CATENIN信号的下调是通过其他途径实现的。
先前的研究表明,DLBCL患者AKT过度激活(39)AKT通过丝氨酸9磷酸化使GSK3β失活,从而促进mTOR信号传导和WNT/β-CATENIN靶基因的翻译(31,32,34). 根据WNT/β-CATENIN和AKT/GSK3β信号之间存在的串扰,我们评估了AXIN2和WIF1对其调节和表达的作用循环蛋白D1,c-MYC公司、和苏维文AXIN2和/或WIF1的异位表达表明,尽管每种拮抗剂单独对AKT/GSK3β信号传导和WNT/β-CATENIN靶基因的表达有中等影响,但它们的共同表达有更显著的影响(图3,一个和B类). 这些结果表明AXIN2和WIF1协同作用使AKT失活,进而导致GSK3β活化和WNT/β-CATENIN靶基因表达下调。巧合的是,当我们测试从淋巴瘤细胞培养基中释放和排泄的WIF1的作用时,我们发现WIF1能够灭活AKT/GSK3β信号并关闭循环蛋白D1,c-MYC公司、和苏维文表达式(图S6和支持实验程序). 然而,当使用特定的WIF1抗体中和WIF1时,其抑制作用被取消,突出了其生物功能的特异性(图S6,B类和D类). 此外,当我们评估AKT失活对WNT/β-CATENIN靶基因表达的影响时,我们发现AKT的抑制足以导致循环蛋白D1,c-MYC公司、和苏维文(图3,D类和E类). 当PRMT5被敲除或抑制时,也观察到类似的结果,尽管与shPRMT5相比,CMP-5对GSK3β去磷酸化/再活化和WNT/β-CATENIN靶基因表达抑制的动力学更快(图4和图S4).
在PRMT5活性存在和不存在的情况下,分析不同患者衍生淋巴瘤细胞系中AXIN2和WIF1的蛋白表达谱,表明PRMT5促进淋巴瘤细胞生长和存活的机制在不同淋巴瘤细胞类型之间是保守的。此外,当我们检测NHL临床样本中WNT/β-CATENIN拮抗剂和下游靶基因的mRNA和蛋白水平时(图7)和小鼠原发性淋巴瘤细胞(图8),我们发现它们的表达与患者源性细胞系的表达具有相同的负相关。此外,β-CATENIN(Ser-675)、AKT(Thr-450/Ser-473)和GSK3β(Ser-9)存在过度磷酸化,这与我们对三种不同淋巴瘤细胞类型的发现一致。淋巴瘤细胞系、临床标本和小鼠原发性淋巴瘤细胞中PRMT5的过度表达与AKT/GSK3β和WNT/β-CATENIN信号的过度激活以及AKT/GSKβ磷酸化和CYCLIN D1、c-MYC的丢失呈正相关,PRMT5敲除或抑制后的SURVIVIN表达表明,PRMT5控制着相互作用的生长调节通路的复杂网络。通过抑制AXIN2型和WIF1系列,PRMT5能够促进AKT/GSK3β信号传导,从而显著增强CYCLIN D1、c-MYC和SURVIVIN的转录和翻译。
通过组蛋白修饰调节染色质结构是基因转录过程中核小体DNA或多或少可访问的主要机制之一。当转位到细胞核时,β-CATENIN与转录因子TCF的一个成员复合,并向WNT/β-CATEN靶基因的启动子区域招募许多转录共激活物。我们已经监测了三种不同淋巴瘤细胞系中所有三个WNT/β-CATENIN靶基因的启动子区与联合激活物和联合阻遏物的结合(图6和图S5)以及小鼠原发性淋巴瘤细胞(图8,E类和F类). 我们已经表明,在存在PRMT5的情况下,组蛋白乙酰转移酶、CBP和p300以及组蛋白H3K4特异性甲基转移酶MLL1的招募占优势。这与组蛋白H3K9/K14乙酰化和H3K4甲基化的富集水平一致,这两种水平已知与激活的转录有关。然而,当PRMT5被敲除时,共激活物的招募丢失,取而代之的是联合阻遏物HDAC2和LSD1的强化招募,它们分别能去乙酰化和去甲基化组蛋白H3标记。
为了进一步了解共激活物和共阻遏物的差异募集是如何受到影响的,我们测量了β-CATENI在细胞启动子区的募集循环蛋白D1,c-MYC公司、和苏维文在正常B细胞和淋巴瘤细胞系中(图5一个). 出乎意料的是,我们发现尽管淋巴瘤细胞系中WNT/β-CATENIN靶基因启动子区的β-CATEN募集增加,但当PRMT5被敲除或抑制时,其结合没有改变(图5,B类和C类). 可以想象,所有三个WNT/β-CATENIN靶基因启动子区发生的动态变化是由于AKT介导的MLL1和或CBP/p300的直接磷酸化所致。或者,AKT可能导致包括MLL1和CBP/p300在内的共激活物复合体内的亚单位磷酸化。有证据支持这两种情况;例如,以前的研究表明,视网膜母细胞瘤结合蛋白5(RbBP5)的磷酸化可能是PI3K/AKT刺激MLL1活性的途径,该蛋白是含有MLL1的COMPASS复合物的一种成分(40). 在另一项研究中,使用激素诱导的前列腺癌大鼠模型,证明PI3K/AKT信号激活MLL1,进而导致H3K4甲基化和32个基因的上调(41). 对这种情况下MLL1启动机制的分析表明,当PI3K/AKT激活时,MLL1经历Taspase-1依赖性裂解成N-和C-末端片段,这些片段与MLL1的催化活性形式有关编号320/C180更有趣的是,对H3K4三甲基化的评估表明,该标记在转录起始点附近富集,用于涉及信号转导、细胞周期调节和肿瘤发生的几个靶基因。这与我们的结果一致,我们的结果表明,AKT在淋巴瘤细胞系、临床样本和表达PRMT5水平升高的小鼠原发性淋巴瘤细胞中被激活,MLL1的募集和其诱导的H3(Me三)启动子区WNT/β-CATENIN靶基因中K4标记增加(图6和图S5). 然而,当PRMT5被抑制或击倒时,AKT变得不活跃,导致MLL1招募和H3(Me三)基因启动子区K4甲基化循环蛋白D1,c-MYC公司、和苏维文需要进行更详细的研究,这超出了本研究的范围,以确定AKT是否通过Taspase-1介导的裂解或通过淋巴瘤细胞MLL复合体内的亚单位磷酸化激活MLL1。
AKT和CBP/p300之间的相互作用已被证明,很明显,由于其在苏氨酸1871上的AKT介导的磷酸化,导致H3K18乙酰化和靶基因失活的减少,结果并不总是阳性的,如CBP失活所示(42). 然而,PI3K/AKT信号也被证明可以促进Smad3和CBP之间的联系,并增强其CBP介导的乙酰化作用,从而导致靶基因激活(43). 此外,AKT对丝氨酸1384上p300的磷酸化已被证明可以增强其乙酰转移酶活性,并导致底物乙酰化增加(44). 由于p300可以作用于组蛋白和非组蛋白,很明显,所有下游靶蛋白都会受到影响,p300介导的ADA3(激活3的改变/缺乏)乙酰化证明了这一点,它会刺激细胞周期进展和抑制凋亡(44). p300共同激活基因表达的另一个机制是通过其启动子组蛋白的直接乙酰化。虽然我们不能排除CBP和p300乙酰化通过β-CATENIN募集而组装的转录复合物中的其他成分的可能性,但我们的研究结果表明,在PRMT5存在下,CBP/p300的结合增加与组蛋白H3K9和K14乙酰化增强之间存在正相关(图6). 我们还观察到,在PRMT5被抑制或击倒后,AKT失活和CBP/p300募集的丢失都出现在循环蛋白D1,c-MYC公司、和苏维文组蛋白和非组蛋白磷酸化和乙酰化的这些变化可能为拮抗酶如HDAC2和LSD1被招募到WNT/β-CATENIN靶基因的启动子区域提供了机会,并将表观遗传景观从转录活性形式改变为非活性形式。因此,PRMT5激活参与调节细胞生长和增殖的主要激酶的能力,从而可以调节其他表观遗传程序的活性,这是这种多功能精氨酸甲基转移酶重编程癌细胞表观基因组的另一种方式。
实验程序
质粒构建
PRMT5击倒如前所述(12)使用慢病毒构建物表达对照GFP(pLenti X2 DEST/shGFP)、shPRMT5-1(pLenti X2 DEST/shPRMT5-1)(用于感染患者衍生淋巴瘤细胞系)或GFP-shPRMT-5-1(pLente-CMV-GFP-DEST/shPRMT5-2),用于感染小鼠原发性Eμ-BRD2淋巴瘤细胞。质粒pLenti X2 DEST/shPRMT5-1和pLenti-CMV-GFP-DEST/shPRMT 5-1使用正义(5′-GATCCCGCCCAGTTTAGATGCTTATGTGTTCCATAAGGCACATGGTTGGAAA-3′)和反义(5’-AGCTTCCAAAAGCCCAGGTGATGGCCTTATGCAGCACACACACATAAGGCGTGG-3′)寡核苷酸构建,其设计包括与BglII和HindIII兼容的粘性末端,并被克隆到BglII–HindIII线性化pENTER/pTER中+矢量。阳性克隆通过EcoRI或EcoRV消化鉴定,并通过DNA测序确认。接下来,质粒pENTER/pTER+/如前所述,shPRMT5-1分别重组为pLenti X2 DEST或pLenti-CMV-GFP-DEST载体(12,45)在pLenti X2 DEST/shPRMT5-1的情况下,通过EcoRV消化或在pLenti-CMV-GFP-DEST/sh PRMT5的情况下通过EcoRI消化鉴定阳性克隆,然后进行DNA测序。质粒pCMV6-entry、pCMV6-entry-AXIN2和pCMV-entry-WIF1购自马里兰州Rockville的Origene Technologies,Inc。
细胞培养、B细胞分离、感染和转染分析
患者来源的淋巴瘤细胞系和小鼠原发性Eμ-BRD2淋巴瘤细胞在补充有10%FBS和1mL的RPMI 1640培养基中生长米丙酮酸钠。如前所述,从全国儿童医院通过合作人类组织网络获得的扁桃体组织中分离出正常的人类B细胞(10,11). 为了激活静止的B细胞,纯化的B淋巴细胞以5×10的密度重新悬浮6细胞/ml在含有10%FBS的RPMI 1640培养基中,然后通过添加15 ng/ml重组人白细胞介素-4(Thermo Fisher Scientific,Waltham,MA)和15μg/ml山羊抗人IgG+IgM(Jackson ImmunoResearch Laboratories,Inc.,West Grove,PA)诱导细胞进入细胞周期。为了产生表达控制性shGFP或shPRMT5 RNA的慢病毒颗粒,将15μg控制质粒pLenti X2 DEST/shGFP、质粒pLenti X2 DEST/shPRMT5-1或pLenti-CMV-GFP-DEST/shPRMT15连同15μg pLP1、6μg pLP 2和3μg pVSG转染到5×106293T细胞,转染前1天接种。转染后约72小时,收获对照shGFP或shPRMT5慢病毒上清液,通过2000rpm离心4分钟澄清,并用于感染患者来源的淋巴瘤细胞系或小鼠原代Eμ-BRD2淋巴瘤细胞,如前所述(12). 简而言之,1×105淋巴瘤细胞在含有10%FBS的RPMI 1640培养基中重新悬浮,并感染400μl shGFP或shPRMT5慢病毒上清液,该上清液预先与10μg Polybrene(Sigma)孵育。感染后72小时制备总RNA、全细胞提取物和交联染色质。
为了确定PRMT5抑制对WNT/β-CATENIN拮抗剂和靶基因表达的影响,用等量(8×105)用对照DMSO或CMP-5(50μ米)处理48小时后制备总RNA、全细胞提取物和交联染色质。为了评估AXIN2和WIF1再表达对AKT/GSK3β和WNT/β-CATENIN信号传导的影响,在电穿孔成1×10之前,将~2μg质粒pCMV6-entry、pCMV6-entry-AXIN1、pCMV-entry-AXIN2和pCMV-entry-WIF1与100μl Amaxa细胞系Nucleofector Kit V(目录号VCA-1003)混合6制造商指定的淋巴瘤细胞(Lonza Co.,Allendale,NJ)。转染72小时后制备总RNA和全细胞提取物。
细胞增殖和荧光激活细胞分选(FACS)
为了评估PRMT5抑制对淋巴瘤细胞生长和增殖的影响,将相同数量的淋巴瘤细胞(8×105)用对照DMSO或CMP-5(50μ米)用台盼蓝染色法每2天计数一次活细胞,共6天。为了确定发生凋亡的淋巴瘤细胞的百分比,对照未经治疗的淋巴瘤细胞或经二甲基亚砜或CMP-5治疗过的淋巴瘤细胞在治疗后72小时采集,并按照制造商(BD Biosciences)的规定用抗膜联蛋白V抗体和碘化丙啶染色。接下来,用Beckman Coulter FC500流式细胞仪对样本进行分析。
淋巴瘤临床样本和动物研究
所有使用无患者识别码的患者淋巴瘤样本的研究均遵守赫尔辛基原则声明,并经俄亥俄州立大学综合癌症中心机构审查委员会(IRB协议编号1997CO194)批准,并按照批准的指南(IBC协议编号2006R0017-R1-AM6)进行同样,所有动物研究均按照联邦和俄亥俄州立大学动物护理和使用委员会批准的指南(IACUC协议编号:2009A0094-R3)进行。
RT-PCR和ChIP分析
使用TRIzol试剂(Thermo Fisher Scientific,Waltham,MA)分离总RNA,并按照前面所述进行实时RT-PCR(12). 如前所述,为了测量目标基因的mRNA水平,使用Applied Biosystems TaqMan分析在10μl反应中进行实时PCR(10). 为了使mRNA水平正常化18秒用1×预先混合的方法在对照和试验细胞系中测量rRNA18秒引物/探针组(加利福尼亚州福斯特市应用生物系统公司)。为了监测靶基因的招募,使用来自正常或转化B细胞的交联染色质进行ChIP分析,当进行PRMT5抑制时,如前所述,从未处理细胞或处理淋巴瘤细胞制备交联染色质(12). 用琼脂糖凝胶电泳分析染色质,以确保DNA片段大小不超过500 bp,并用30μl蛋白A磁珠预先清除染色质样品,这些磁珠用封闭缓冲液(0.2 mg/ml剪切鲑鱼精子DNA,0.5 mg/ml BSA)处理过夜在与前免疫或免疫抗体孵育之前。实时RT-PCR分析中使用的引物集和探针列于表1中列出了实时ChIP分析中使用的引物集和探针表2.
表1。
| 底漆 |
正向序列 |
反转顺序 |
探查 |
| PRMT5项目 |
5′-TATGTGTACGGCTCACA3-′ |
5′-TGGCTGAAGGTGAAAACAGG3-′ |
31 |
| AXIN1型 |
5′-AGCTCTCCGAGACAGAGACAGACAA-3′ |
5′-ACAACGATGCTGTCACACG-3′ |
6 |
| AXIN2型 |
5′-GCTAGAGTGCGTTCATGGT-3′ |
5′-AGGACGTGTGCAAAGCAT-3′ |
8 |
| WIF1系列 |
5′-CCAGGGAGCTCTCAA-3′ |
5′-TTGGTTCATGGCAGGTT-3′ |
76 |
| β-卡特宁
|
5′-TTAAAAAAGCCAGTTTGGGTAAAAA-3′ |
5′-CCCACCCTACACAAGT-3′ |
13 |
| β-肌动蛋白
|
5′-GGTAGACGCGATCTGG-3′ |
5′-GGCATGGAATCAACCACCTCAAC-3′ |
6 |
| 循环蛋白D1 |
5′-GAAGATCGTCGCCACCTG-3′ |
5′-GACCTCTCCTCGCACTTCT-3′ |
67 |
| c-MYC公司 |
5′-CACCAGCAGACTCTGA-3′ |
5′-GATCCAGACTCTGACTTTTG-3′ |
34 |
| 苏维文 |
5′-GCCAGTGTTCTTCTGCTT-3′ |
5′-CCGGACGAATGCTTTTATG-3′ |
11 |
| AKT公司 |
5′-GGCTATTGTGAAGGAGGGTTG-3′ |
5′-TCCTTGTAGCCAATGAAGGTG-3′ |
69 |
|
葛兰素史克3β |
5′-GACATTTCACCTCAGAGTGCGC-3′ |
5′-GTTTAGTCGGCAGTTGGTGT-3′ |
67 |
| 项目5 |
5′-GCTGTCACCTGAGTGTCTGG-3′ |
5′-GATGCTCACGCCATCATCT-3′ |
99 |
| Axin1轴 |
5′-GGGCCCCCTCAAGTAGAC-3′ |
5′-CCCTCCAAGATCCATACCTG-3′ |
13 |
| 轴2 |
5′-CTGGTGTGGTCGCTACAG-3′ |
5′-TGACACTGCTGATGGTGTAG-3′ |
77 |
| 无线1 |
5′-ttaagtgagaggcgtgtgtcg-3′ |
5′-GCAGACACTGCAAGAGG-3′ |
40 |
| β-连环蛋白
|
5′-TTCCTATGGGAACAGTCGAAG-3′ |
5′-TTGTATTTACTCCTACAA-3′ |
93 |
| β-肌动蛋白
|
5′-GCAAAACATCCCAAATT-3′ |
5′-TTTTCATGGATACTTGGAATGACTA-3′ |
5 |
| 细胞周期蛋白D1 |
5′-CTCTCTTCGCACTTCGCT-3′ |
5′-GAGATGTGCACATCCATGC-3′ |
67 |
| c-Myc公司
|
5′-TCTTCCCTCATCTTCTTGCTCTTC-3′ |
5′-CCTAGTGCTGCATGAGGAGA-3′ |
77 |
| Survivin公司 |
5′-CCTGGCCCAGATACTA-3′ |
5′-GGACTTAGGGAACAAGGAACC-3′ |
56 |
|
Gsk3型β |
5′-CAAGAGAGCCATGTCG-3′ |
5′-TGGTTACCTCGCTGCCATCT-3′ |
10 |
表2。
| 底漆 |
正向序列 |
反转顺序 |
探查 |
| AXIN1型 |
5′-ACTCCTGGGAGCTCTT-3′ |
5′-CAGTACATCGGAGGGCAGTC-3′ |
81 |
| AXIN2型 |
5′-TCTTGCCTTCCTCTCACCTT-3′ |
5′-CACACTTTTCGGGGTTGG-3′ |
24 |
| WIF1系列 |
5′-CGGAGGGTCAGGTACAGCTA-3′ |
5′-CCCTGATAAACCCGGCAGT-3′ |
81 |
| 循环蛋白D1 |
5′-CGGCTTTTGATCTTGCTTA-3′ |
5′-TCTGCTGCTCGCTGCTACT-3′ |
1 |
| c-MYC公司
|
5′-GAATTGGGAACTCGTGTG-3′ |
5′-CTAGGGCGAGGGGTT-3′ |
53 |
| 苏维文 |
5′-AAGGAGAGTTTGCCCTGAG-3′ |
5′-GGGCCACTACGCTGATAAGA-3′ |
24 |
| 细胞周期蛋白D1 |
5′-CTAGCTGTCCTCCTGTCCAGA-3′ |
5′-GGGCTTCTCTCCCTAAGAGG-3′ |
10 |
| c-Myc公司
|
5′-TCATGCTGCGCTATTACTGTTT-3′ |
5′-CCTCTGCTTTGGGAACTCG-3′ |
63 |
| Survivin公司 |
5′-GGAGGACCCTCTTAGGGAAA-3′ |
5′-AATCGCTCGGACTGCTTCT-3′ |
48 |
ChIP-Seq图书馆建设、数据处理和分析
对于ChIP-Seq分析,交联染色质由5×10制备6正常B、Pfeiffer或SUDHL-2细胞被免疫抗H3(Me)免疫沉淀2)R8,之前已经描述过(10,17). 对于所有类型的细胞,按照前面所述进行ChIP分析(10,11). 使用来自两个不同批次细胞的交联染色质在不同培养日进行重复的ChIP-Seq实验。在开始构建文库之前,检查免疫沉淀DNA是否在两个已建立的PRMT5靶启动子上富集H3R8对称甲基化,ST7标准和RBL2型,单位:ChIP与使用实时PCR输入样本。接下来,使用Illumina Hi-Seq 2000 Prep Kit(Illuminia,Inc.,圣地亚哥)创建库。在适配器连接步骤后,通过琼脂糖凝胶电泳选择大小在300到400 bp之间的DNA片段,然后进行15个扩增周期。再次进行实时PCR以确认对照物的富集ST7标准和RBL2型启动子靶序列。然后使用高通量Illumina GAIx测序仪对ChIP-Seq DNA文库进行测序,并使用Eland管道(Illumina,Inc.,San Diego)将DNA序列读数与UCSC人类基因组组装HG18比对。为了进行分析,抗H3(Me2)使用来自正常和转化B细胞的R8 ChIP-Seq数据来确定PRMT5的全基因组结合位点。首先,DNA序列读取与人类参考基因组(GRCh37)对齐,Rsubread版本1.22.2(46),副本用Picard工具版本1.94标记(http://broadinstitute.github.io/picard网站/),三使用MACS2 2.1.1版执行峰值调用(47). 使用NGSplot工具,使用一系列内部shell和R脚本生成转录起始位点周围的一致峰值和读取计数(48),基因组范围(49)和ChIPpeakAnno(50)包。
抗体和免疫印迹分析
如前所述进行蛋白质提取、免疫印迹和免疫沉淀(10,12,51). 简单地说,在RIPA缓冲液(50 m)中制备全细胞提取物米三氯化氢,pH 7.5,150 m米NaCl,1%Nonide P-40,0.5%脱氧胆酸钠,0.5 m米DTT,1米米苯甲基磺酰氟)补充蛋白酶和磷酸酶抑制剂(10μg/ml盐酸苯甲脒、10μg/ml抑肽酶、10μg/ml亮氨酸蛋白酶、2.25μg/ml胃蛋白酶抑制素A、10 m米β-甘油磷酸,1米米原钒酸钠,50 m米氟化钠)。接下来,在8–12%SDS-PAGE上分离蛋白质,转移到聚偏二氟乙烯膜上,并使用Amersham Biosciences ECL Western印迹检测试剂(GE Healthcare)通过增强化学发光进行检测。对于蛋白质检测,如前所述,使用BRG1、PRMT5及其表观遗传标记的抗体(10). 抗CBP(sc-7300)、p300(sc-48343)、CYCLIN D1(sc-718)、c-MYC(sc-40)和WIF1(sc-25520(目录号9267)和抗p-AKT(Ser-473)(目录号4060)抗体购自马萨诸塞州丹佛市Cell Signaling Technology;和抗AXIN1(ab131372)、抗AXIN2(ab32197)、抗H3(Me三)K4(ab8580),抗H3(Me三)K9(ab8898),抗H3(Me三)K27(ab6002)、抗LSD1(ab17721)、抗JMJD6/UTX(ab50720)和抗HDAC2(ab32117)抗体购自英国剑桥Abcam。抗H3K9(Ac)(07-352)和抗H3K14(Ac,抗β-肌动蛋白(SAB5500001)抗体购自Sigma。
统计分析
所有实时RT-PCR、ChIP和免疫印迹实验使用不同样品至少进行三次,结果表示为平均值±S.D.配对t吨Sidak多重比较调整后的方差检验与分析t吨测试用于生成第页两个样本之间的比较值。
作者贡献
J.C.和V.K.形式分析;J.C.、V.K.和S.S.验证;J.C.和V.K.调查;J.C.、V.K.和S.S.可视化;R.A.B.和S.S.资源;R.A.B.和S.S.监督;R.A.B.和S.S.资金收购;S.S.概念化;S.S.书面原稿;S.S.项目管理;S.S.写作审查和编辑;S.准备了手稿的初稿,所有合著者都对其进行了审查,并准备了手稿和数字的定稿。
致谢
我们感谢A.N.Imbalzano提供慢病毒表达载体,感谢G.Denis善意地提供小鼠原发性Eμ-BRD2淋巴瘤细胞及其使用建议,感谢O.H.Gulcin帮助进行ChIP-Seq数据分析。
这项工作得到了白血病和淋巴瘤学会奖MCL7001-18(授予R.A.B.)和国家研究基金(卡塔尔基金会成员)国家优先研究计划拨款NPRP8-617-3-131(授予S.S)的支持。作者声明,他们与本文的内容没有利益冲突.
三请注意,JBC不负责本网站或任何其他第三方托管网站的长期存档和维护。
2使用的缩写如下:
- MCL公司
套细胞淋巴瘤
- 非霍奇金淋巴瘤
非霍奇金淋巴瘤
- DLBCL(DLBCL)
弥漫性大B细胞淋巴瘤
- GCB公司
生发中心B细胞
- 基础知识
活化B细胞样
- TCF公司
T细胞因子
- LEF公司
淋巴增强因子
- FBS公司
胎牛血清
- 圆周率
预先免疫
- 第页
短发夹
- PI3K系列
磷脂酰肌醇3-激酶。
工具书类
-
1Kuppers R.、Klein U.、Hansmann M.L.和Rajewsky K.(1999)人类B细胞淋巴瘤的细胞起源。北英格兰。《医学杂志》3411520–152910.1056/NEJM19991111342007[内政部] [公共医学] [谷歌学者]
-
2Stevenson F.K.、Sahota S.S.、Ottensmeier C.H.、Zhu D.、Forconi F.和Hamblin T.J.(2001)B细胞源性人类恶性肿瘤中V基因突变的发生和意义。高级癌症研究83,81–11610.1016/S0065-230X(01)83004-9[内政部] [公共医学] [谷歌学者]
-
三。Nogai H.、Dörken B.和Lenz G.(2011)非霍奇金淋巴瘤的发病机制。临床杂志。昂科尔。29, 1803–181110.1200/JCO.2010.3252[内政部] [公共医学] [谷歌学者]
-
4《世界卫生组织造血和淋巴组织肿瘤分类》中的Swerdlow S.H.、Campo E.、Seto M.和Müller-Hemrelink H.K.(2008),第229-232页。IARC出版集团,法国里昂[谷歌学者]
-
5Alizadeh A.A.、Eisen M.B.、Davis R.E.、Ma C.、Lossos I.S.、Rosenwald A.、Boldrick J.C.、Sabet H.、Tran T.、Yu X.、Powell J.I.、Yang L.、Marti G.E.、Moore T.、Hudson J.Jr.等人(2000)通过基因表达谱确定的不同类型的弥漫性大B细胞淋巴瘤。自然403, 503–51110.1038/35000501[内政部] [公共医学] [谷歌学者]
-
6Rosenwald A.,Wright G.,Chan W.C.,Connors J.M.,Campo E.,Fisher R.I.,Gascoyne R.D.,Muller-Hermelink H.K.,Smeland E.B.,Giltnane J.M..,Hurt E.M.,Zhao H.,Averett L.,Yang L.等人(2002年)《利用分子剖析预测弥漫性大B细胞淋巴瘤化疗后的生存率》。北英格兰。《医学杂志》3461937-194710.1056/NEJMoa012914[内政部] [公共医学] [谷歌学者]
-
7Pezzella F.、Gatter K.C.和Mason D.Y.(1989)《14例检测》;石蜡包埋淋巴瘤组织中的18染色体易位。柳叶刀1, 779–780[内政部] [公共医学] [谷歌学者]
-
8MotllóC.、Grau J.、JuncaáJ.、Ruiz N.、Mate J.L.、Orna E.、Navarro J.T.、Vives S.、Sancho J.M.、Esteban D.、Granada I.、Feliu E.、Ribera J.和MilláF.(2010)两例三重命中淋巴瘤的转移(3;8)(q27;q24)。癌症遗传学。细胞遗传学。203、328–3322016年10月10日/j.cancergencyto.2010.08.018[内政部] [公共医学] [谷歌学者]
-
9Torres C.H.和Tirado C.A.(2018)两例双发淋巴瘤:分子细胞遗传学方法。遗传学协会杂志。Technol公司。44, 141–145[公共医学] [谷歌学者]
-
10Pal S.、Baiocchi R.A.、Byrd J.C.、Grever M.R.、Jacob S.T.和Sif S.(2007)低水平的miR-92b/96诱导套细胞淋巴瘤中PRMT5翻译和H3R8/H4R3甲基化。EMBO期刊26,3558–356910.1038/sj.emboj.7601794[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
11Wang L.、Pal S.和Sif S.(2008)蛋白质精氨酸甲基转移酶5抑制白血病和淋巴瘤细胞中肿瘤抑制因子RB家族的转录。分子细胞。生物学28,6262–627710.1128/MCB.00923-08号[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
12Chung J.、Karkhanis V.、Tae S.、Yan F.、Smith P.、Ayers L.W.、Agostinelli C.、Pileri S.、Denis G.V.、Baiocchi R.A.和Sif S.(2013)《蛋白精氨酸甲基转移酶5(PRMT5)抑制通过视网膜母细胞瘤抑癌途径的重新激活和多梳阻遏物复合物2(PRC2)沉默诱导淋巴瘤细胞死亡》。生物学杂志。化学。288、35534–3554710.1074/jbc。M113.510669型[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
13Alinari L.、Mahasenan K.V.、Yan F.、Karkhanis V.、Chung J.H.、Smith E.M.、Quinion C.、Smiths P.L.、Kim L.、Patton J.T.、Lapalonbella R.、Yu B.、Wu Y.、Roy S.、De Leo A.等(2015)蛋白质精氨酸甲基转移酶5的选择性抑制阻止B细胞转化的启动和维持。血液125, 2530–254310.1182/血液-2014-12-619783[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
14Karkhanis V.、Hu Y.J.、Baiocchi R.A.、Imbalzano A.N.和Sif S.(2011)PRMT5诱导甲基化在生长控制和发育中的多功能性。生物化学趋势。科学。36, 633–64110.1016/j.tibs.2011.09.001[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
15Yang Y.和Bedford M.T.(2013)蛋白质精氨酸甲基转移酶与癌症。Nat.Rev.癌症13, 37–5010.1038/编号3409[内政部] [公共医学] [谷歌学者]
-
16Shailesh H.、Zakaria Z.Z.、Baiocchi R.和Sif S.(2018)《癌症中的蛋白质精氨酸甲基转移酶5(PRMT5)失调》。Oncotarget公司9, 36705–3671810.18632/目标26404[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
17Pal S.、Vishwanath S.N.、Erdjument-Bromage H.、Tempst P.和Sif S.(2004)《人类SWI/SNF-相关PRMT5甲基化组蛋白H3精氨酸8》,负调控ST7和NM23抑癌基因的表达。分子细胞。生物24,9630–964510.1128/MCB.24.21.9630-9645.2004年[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
18Jansson M.、Durant S.T.、Cho E.C.、Sheahan S.、Edelmann M.、Kessler B.和La Thangue N.B.(2008)精氨酸甲基化调节p53反应。自然细胞生物学。10, 1431–143910.1038/ncb1802[内政部] [公共医学] [谷歌学者]
-
19Cho E.C.、Zheng S.、Munro S.、Liu G.、Carr S.M.、Moehlenbrink J.、Lu Y.C.、Stimson L.、Khan O.、Konietzny R.、McGouran J.、Coutts A.S.、Kessler B.、Kerr D.J.和Thangue N.B.(2012)精氨酸甲基化控制E2F-1的生长调节。恩博,1785年至1797年10.1038/emboj.2012.17[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
20Logan C.Y.和Nusse R.(2004)发育和疾病中的Wnt信号通路。每年。Rev.细胞发育生物学。20, 781–81010.1146/annurev.cellbio.20.010403.113126[内政部] [公共医学] [谷歌学者]
-
21Willert K.和Jones K.A.(2006)Wnt信号:党是核心吗?基因发育20,1394–140410.1101/gad.1424006[内政部] [公共医学] [谷歌学者]
-
22Klaus A.和Birchmeier W.(2008)Wnt信号传导及其对发育和癌症的影响。Nat.Rev.癌症8, 387–39810.1038/nrc2389[内政部] [公共医学] [谷歌学者]
-
23Angers S.和Moon R.T.(2009)Wnt信号转导中的邻近事件。自然修订版分子细胞生物学。10, 468–47710.1038/编号2717[内政部] [公共医学] [谷歌学者]
-
24Lai S.L.、Chien A.J.和Moon R.T.(2009)Wnt/Fz信号与细胞骨架:在肿瘤发生中的潜在作用。细胞研究19,532–54510.1038/cr.2009.41[内政部] [公共医学] [谷歌学者]
-
25Mosimann C.、Hausmann G.和Basler K.(2009),β-连环蛋白打击染色质:Wnt靶基因激活的调节。自然修订版分子细胞生物学。10, 276–28610.1038/编号2654[内政部] [公共医学] [谷歌学者]
-
26Niehrs C.(2012)WNT受体信号传导的复杂世界。自然修订版分子细胞生物学。13, 767–77910.1038/个3470[内政部] [公共医学] [谷歌学者]
-
27Henderson B.R.(2000)APC的核质穿梭调节β-catenin亚细胞定位和周转。自然细胞生物学。2, 653–66010.1038/35023605[内政部] [公共医学] [谷歌学者]
-
28Valenta T.、Hausmann G.和Basler K.(2012)β-catenin的多种面孔和功能。EMBO期刊31,2714–273610.1038/emboj.2012.150[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
29Li Y.、Chitnis N.、Nakagawa H.、Kita Y.、Natsugoe S.、Yang Y.、Li Z.、Wasik M.、Klein-Szanto A.J.、Rustgi A.K.和Diehl J.A.(2015)PRMT5是由多种致癌因素触发的淋巴肿瘤所必需的。癌症发现。5, 288–30310.1158/2159-8290.CD-14-0625[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
30MacDonald B.T.、Tamai K.和He X.(2009)Wnt/β-catenin信号:成分、机制和疾病。开发单元17, 9–262016年10月10日/j.devcel.2009.06.016[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
31Cross D.A.、Alessi D.R.、Cohen P.、Andjelkovich M.和Hemmings B.A.(1995)通过蛋白激酶B介导的胰岛素抑制糖原合成酶激酶-3。自然378, 785–78910.1038/378785a0[内政部] [公共医学] [谷歌学者]
-
32Fukumoto S.、Hsieh C.M.、Maemura K.、Layne M.D.、Yet S.F.、Lee K.H.、Matsui T.、Rosenzweig A.、Taylor W.G.、Rubin J.S.、Perrella M.A.和Lee M.E.(2001)Akt通过Dishevelled参与Wnt信号通路。生物学杂志。化学。276, 17479–1748310.1074/jbc。C000880200号[内政部] [公共医学] [谷歌学者]
-
33Hart J.R.和Vogt P.K.(2011)AKT的磷酸化:突变分析。Oncotarget公司2, 467–47610.18632/目标293[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
34McCubrey J.A.、Steelman L.S.、Bertrand F.E.、Davis N.M.、Abrams S.L.、Montalto G.、D'Assoro A.B.、Libra M.、Nicoletti F.、Maestro R.、Basecke J.、Cocco L.、Cervello M.和Martelli A.M.(2014)GSK-3和Wnt/β-catenin在造血和白血病发生中的多方面作用:治疗干预的机会。白血病28, 15–3310.1038/leu.2013.184[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
35Lenburg M.E.、Sinha A.、Faller D.V.和Denis G.V.(2007)肿瘤特异性和增殖特异性基因表达是小鼠转基因B细胞淋巴瘤的典型表现。生物学杂志。化学。282, 4803–481110.1074/jbc。M605870200型[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
36Lin S.Y.、Yeh K.T.、Chen W.T.、Chen H.C.、Chen S.T.、Chiou H.Y.和Chang J.G.(2004)结直肠癌抑癌基因的启动子CpG甲基化及其与临床特征的关系。昂科尔。代表11、341–348[公共医学] [谷歌学者]
-
37杨丽华,徐华堂,李启才,蒋国勇,张晓平,赵华英,徐坤,王爱华(2013)Axin基因在肺癌中的异常高甲基化及其临床病理意义。肿瘤生物学。34, 749–75710.1007/s13277-012-0604-z[内政部] [公共医学] [谷歌学者]
-
38Thorvaldsen T.E.、Pedersen N.M.、Wenzel E.M.和Stenmark H.(2017)AXIN1和AXIN2在坦克酶抑制剂诱导的降解体形成和β-连环蛋白降解中的差异作用。公共科学图书馆12,e017050810.1371/journal.pone.0170508[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
39Wang J.,Xu-Monette Z.Y.,Jabbar K.J.,Shen Q.,Manyam G.C.,Tzankov A.,Visco C.,Wang J,Montes-Moreno S.,Dybkaer K.,Tam W.,Bhagat G.,His E.D.,van Krieken J.H.,Ponzoni M.等人(2017),AKT过度激活和AKT靶向治疗弥漫性大B细胞淋巴瘤的潜力。美国病理学杂志。187, 1700–171610.1016/j.ajpath.2017.04.009[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
40Zhang P.、Chaturvedi C.P.、Tremblay V.、Cramet M.、Brunzelle J.S.、Skinotis G.、Brand M.、Shilatifard A.和Couture J.F.(2015)RbBP5上的磷酸化开关调节组蛋白H3-Lys4甲基化。基因发育29、123–12810.1101/gad.254870.114[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
41Wang Q.、Trevino L.S.、Wong R.L.、Medvedovic M.、Chen J.、Ho S.M.、Shen J.、Foulds C.E.、Coarfa C.、O'Malley B.W.、Shilatifard A.和Walker C.L.(2016)MLL1表观基因组重编程将早期环境暴露与前列腺癌风险联系起来。分子内分泌。30, 856–8712015-1310年10月21日[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
42Liu Y.,Xing Z.B.,Zhang J.H.和Fang Y.(2013)Akt激酶以CBP与组蛋白H3的结合为靶点,调节赖氨酸18的乙酰化。FEBS信函。587, 847–8532016年10月10日/j.febslet.2013.023[内政部] [公共医学] [谷歌学者]
-
43Das F.、Ghosh-Chudhury N.、Venkatesan B.、Li X.、Mahimainathan L.和Choudhury G.(2008)Akt激酶靶向CBP与SMAD3的关联,以调节TGFβ诱导的纤溶酶原激活物抑制剂-1的表达。J.细胞。生理学。214, 513–52710.1002/jcp.21236[内政部] [公共医学] [谷歌学者]
-
44Srivastava S、Mohibi S、Mirza S、Band H和Band V(2017)表皮生长因子受体激活通过AKT-p300途径促进ADA3乙酰化。细胞周期16, 1515–152510.1080/15384101.2017.1339846[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
45Karkhanis V.、Wang L.、Tae S.、Hu Y.J.、Imbalzano A.N.和Sif S.(2012)蛋白质精氨酸甲基转移酶7通过甲基化聚合酶δ催化亚基基因POLD1的启动子组蛋白H2A和H4调节细胞对DNA损伤的反应。生物学杂志。化学。287, 29801–2981410.1074/jbc。M112.378281号[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
46Liao Y.、Smyth G.K.和Shi W.(2013)Subread校准器:通过seed-and-vote快速、准确和可扩展的读取映射。核酸研究41,e10810.1093/nar/gkt214[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
47Zhang Y.、Liu T.、Meyer C.A.、Eeckhoute J.、Johnson D.S.、Bernstein B.E.、Nusbaum C.、Myers R.M.、Brown M.、Li W.和Liu X.S.(2008)基于模型的ChIP-Seq分析(MACS)。基因组生物学。第9页,第137页10.1186/gb-2008-9-9-r137[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
48Shen L.、Shao N.Y.、Liu X.、Maze I.、Feng J.和Nestler E.J.(2013)diffReps:利用生物复制品检测ChIP-seq数据中的差异染色质修饰位点。公共科学图书馆8,e6559810.1371/日记本.0065598[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
49Lawrence M.、Huber W.、Pagès H.、Aboyoun P.、Carlson M.、Gentleman R.、Morgan M.T.和Carey V.J.(2013)计算和注释基因组范围的软件。公共科学图书馆计算。生物9,e100311810.1371/日记.pcbi.1003118[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
50Zhu L.J.、Gazin C.、Lawson N.D.、Pagès H.、Lin s.M.、Lapointe D.s.和Green M.R.(2010)ChIPpeakAnno:注释ChIP-seq和ChIP-ChIP数据的生物导体包。BMC生物信息学11, 23710.1186/1471-2105-11-237[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
51Tae S.、Karkhanis V.、Velasco K.、Yaneva M.、Erdjument-Bromage H.、Tempst P.和Sif S.(2011)Bromodomain蛋白7与PRMT5和PRC2相互作用,并参与其靶基因的转录抑制。核酸研究39,5424–543810.1093/nar/gkr170[内政部] [PMC免费文章] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。

