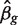简介
DNA测序的成本继续以惊人的速度下降(1)。正如它所做的那样,测序技术作为研究基因表达的平台变得越来越有吸引力。当前的“下一代”测序技术通过生成短读或序列标签来测量基因表达,即35-300个碱基对的序列对应于原始RNA的片段。有许多技术和许多不同的协议。流行的方法是基于标记的方法,包括tag-Seq(2),深SAGE(三)、SAGE-Seq(4)每个基因中一个或多个锚定位置的序列,或RNA-Seq(5——8)对整个转录组中的随机片段进行排序。这两种方法在研究基因表达和调控方面都已证明是成功的(9——11)。在本文中,我们将通用术语RNA-Seq来包括任何基于标记的或RNA-Seq-变体,其中非常高通量的测序应用于RNA片段。
为了评估不同条件下的差异表达,读取计数在感兴趣的基因组水平上进行总结,例如基因或外显子。尽管RNA-Seq可以用于寻找新的外显子或剪接变异和异构体特异性表达(7,12——14),转录汇编(15)或等位基因特异表达(16,17),我们在这篇文章中的重点是预先确定的基因组特征的差异表达。然而,当应用于外显子或外显子连接等亚基因特征时,这里开发的方法与推断异构体特异性差异表达相关。
线性建模方法在分析具有多个解释因素的微阵列实验方面非常成功(18,19)。RNA-Seq用于类似实验已成为一种普遍现象,因此迫切需要能够为复杂RNA-Sq实验提供相同灵活性和严密性的统计方法(20).
RNA-Seq数据分析的一种策略是将读取计数标准化并转换为近似正态,然后分析微阵列数据(三,6,21)。然而,这种方法并没有完全适应读取计数数据的特性。一个问题是,即使在转换后,非常小的计数也远不能正常分布,尽管对于较大的计数,这个问题可以迅速缓解。一个更普遍、更重要的问题是,计数数据通常显示出强大的均值-方差关系,而现有的基于正态分布的分析没有考虑到这一关系,从而导致潜在的低效统计推断。转换,如平方根(三)可以减少但不能完全消除均值-方差相关性。因此,使用适当的分布计算读取计数的准确概率,比简单地转换为正态分布更有可能实现更敏感的统计过程(22——25)。模拟表明,计数模型比近似正常模型具有更强的统计能力来检测差异表达(26)。显式计数模型的另一个优点是,它们为区分生物和技术变异性提供了更精细的可能性(22,23).
尽管测序成本降低,但RNA-Seq实验对许多研究人员来说仍然很昂贵,常常将RNA-Seq-研究限制在少数库中。通常很少有复制。然而,评估与生物变异相关的差异表达的基本科学需求仍未减弱(27)。因此,有必要从极少数的复制文库中尽可能可靠地估计生物变异。不同的基因或转录本可能有不同程度的生物变异,这一事实使问题更加复杂。在微阵列分析中,通过正则化t吨-测试(28)或者更正式地通过经验贝叶斯或基因间共享信息的相关方法(18,19,29).
DNA样本可以被认为是cDNA片段的群体,每个基因组特征可以被认为是要估计群体大小的物种。DNA样本的测序可以被认为是对这些物种中的每一个进行随机取样,目的是估计种群中每一物种的相对丰度。如果每个cDNA片段被选择测序的机会相同,并且片段是独立选择的,那么给定基因组特征的读取计数在相同cDNA样本的重复序列运行中应遵循泊松变化规律。泊松模型表明,平均值等于方差,这一关系已在早期的一项RNA-Seq研究中得到验证,该研究使用了分布在Illumina GA测序器多条通道上的相同初始RNA源(30).
泊松模型没有考虑生物变异性或任何可能导致不同RNA样本之间不同基因相对丰度变化的技术来源。当样本之间的丰度不恒定时,读取计数将相对于泊松过度分散,即方差必须高于平均值。过度离散二项式(31,32)或泊松(32——36)已提出用于基因表达序列分析(SAGE)或RNA-Seq数据的模型。这些建议都不具备在基因之间共享信息的能力,限制了它们进行大量复制库的实验。
在基因之间共享信息的一种非常简单的方法是假设所有基因都遵循相同的均值-方差关系,因此所有具有相同预期计数的基因都具有相同的方差(23,25,37)。这几乎肯定太简单了,因为它不考虑某些基因可能比其他基因更具变异性的可能性。罗宾逊和史密斯(22,24)开发了一种前景看好的经验贝叶斯方法,使用加权似然以基因方式估计生物变异,在Bioconductor包edgeR中实现。针对SAGE提出了其他更明确的贝叶斯方法(38,39)或RNA-Seq数据(40)后者在Bioconductor包baySeq中实现。在模拟数据和实际数据上的比较表明,edgeR和baySeq优于不允许基因特异性变异或不共享基因间信息的替代方法(40)。然而,这些方法仅限于单向布局中各组之间的比较。
对于来自SAGE或RNA-Seq实验的计数数据,建议使用广义线性模型(GLM),计数被视为过分散二项式(31,32,37),泊松(21,41),过分散泊松(32,34)或具有随机效应的泊松(33)。GLM是需要迭代拟合的非线性模型,因此所有这些方法的共同问题是计算时间和某些数据集的某些基因的算法失败。
本文为多因子RNA-Seq实验开发了GLM算法。统计方法是在基因基础上估计生物变异并将其与技术变异分离的。为了使GLM模型拟合更快、更可靠,开发了并行计算方法。为了在基因之间共享信息,开发了一种经验贝叶斯方法,允许基因特定的变异,即使只有少数生物复制可用。该方法为分析任意复杂的RNA-Seq实验提供了一条途径,前提是存在一定程度的生物复制。
材料和方法
生物变异系数
RNA-Seq配置文件由n个RNA样本。设π吉是所有cDNA片段在我-来自基因的第个样本克.让G公司表示基因总数,所以 对于每个样本。让
对于每个样本。让 表示π的变异系数(CV)(标准偏差除以平均值)吉在复制品之间我。我们表示库中映射读取的总数我通过N个我和映射到克-第th个基因年吉.然后
表示π的变异系数(CV)(标准偏差除以平均值)吉在复制品之间我。我们表示库中映射读取的总数我通过N个我和映射到克-第th个基因年吉.然后
假设计数年吉相同RNA样品重复测序遵循泊松分布,混合分布方差的一个众所周知的公式意味着:
将两边分开 给予
给予
第一项1/μ吉是泊松分布的平方CV,第二个是未观察到的表达式值的平方CV。总CV2因此是技术简历2其中π吉测量值加上生物CV2真π的吉。在本文中,我们将其称为克分散度和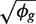 严格地说,生物变异系数捕获了复制品之间的系间变异的所有来源,包括可能来自技术原因的贡献,例如文库准备以及样品之间的真正生物变异。
严格地说,生物变异系数捕获了复制品之间的系间变异的所有来源,包括可能来自技术原因的贡献,例如文库准备以及样品之间的真正生物变异。
广义线性模型
GLM是经典线性模型对非正态分布响应数据的扩展(42,43)。GLM根据它们的均值-方差关系指定概率分布,例如上面为读取计数指定的二次均值-方差的关系。假设估计值可用于克,因此可以计算以下任意值的方差 μ吉,GLM理论可用于拟合对数线性模型
对于每个基因(32,41)。在这里x个我是一个协变量向量,用于指定应用于RNA样本的处理条件我、和β克是一个回归系数向量,通过该向量为基因介导协变量效应克二次方差函数指定负二项GLM分布族。负二项分布的使用等价于处理π吉伽马分布。
装配GLM
对数似然对系数β的导数克是X(X)T型z(z)克,其中X(X)是带列的设计矩阵x个我和z(z)吉 = (年吉 − μ吉)/(1 + ϕ克μ吉)。系数的Fisher信息矩阵可以写成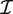 克 = X(X)T型W公司克X(X),其中W公司克是标准GLM理论中工作重量的对角矩阵(43)。求β的最大似然估计的Fisher评分迭代克因此是
克 = X(X)T型W公司克X(X),其中W公司克是标准GLM理论中工作重量的对角矩阵(43)。求β的最大似然估计的Fisher评分迭代克因此是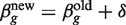 带有δ = (X(X)T型W公司克X(X))−1
X(X)T型z(z)克此迭代通常会增加似然函数,但似然也会降低,表示与所需解的背离。另一方面,始终存在一个步长修饰符α为0 < α < 1这样
带有δ = (X(X)T型W公司克X(X))−1
X(X)T型z(z)克此迭代通常会增加似然函数,但似然也会降低,表示与所需解的背离。另一方面,始终存在一个步长修饰符α为0 < α < 1这样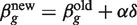 增加了可能性。选择α以便在每次迭代时都是这样,这就是所谓的行搜索策略(44,45).
增加了可能性。选择α以便在每次迭代时都是这样,这就是所谓的行搜索策略(44,45).
Fisher的打分迭代可以看作是一种近似的Newton-Raphson算法,Fisher信息矩阵近似于二阶导数矩阵。线搜索策略可以与正定的二阶导数阵的任何近似值一起使用。我们的实现使用了一个计算方便的近似值。在不损失通用性的情况下,线性模型可以参数化,以便X(X)T型X(X) = 我。如果完成此操作,并且如果 μ吉也恰好是恒定的我对于给定的基因克,则信息矩阵相当简化为 μ克/(1 + ϕ克μ克)乘以单位矩阵我将其作为信息矩阵的近似值,带线搜索修改的Fisher评分步骤变为简单的δ = αX(X)T型z(z)克,其中乘数 μ克/(1 + ϕ克μ克)已被吸收到步长因子α中。在这个公式中,α不再被限制为小于1。在我们的实现中,每个基因都有自己的步长α,随着迭代的进行,步长α会增加或减少。
仿真
使用负二项分布计数生成了固定总数为10的人工数据集 000个基因。根据形状参数为0.5、特别的选择模拟肿瘤数据的大小分布。平均分散度设置为0.16(BCV = 0.4). 在一次模拟中,所有基因都具有相同的真实分散性。在另一个模拟中,根据具有20个自由度的反向chisquare分布,在0.16左右随机生成真实分散度。
结果
技术和生物变异
RNA-Seq实验的起点是一组n个RNA样本,通常与各种治疗条件有关。对每个样本进行测序,将短读映射到适当的基因组,并记录映射到每个感兴趣的基因组特征的读数。为了简化术语,我们将在本文中假设计数是在基因水平上总结的,尽管在实践中基因组特征也可能是转录本、外显子、SAGE标签、外显子连接或非编码RNA。从样本中读取的次数我映射到基因克将被表示年吉.样本的基因计数集我组成表达式配置文件或图书馆对于该样本。每个计数的预期大小是库大小和该样本中该基因的相对丰度的乘积。
在任何RNA-Seq实验中都可以区分两个水平的变异。首先,每个基因的相对丰度在不同的RNA样本中会有所不同,主要是由于生物原因。其次,存在测量误差,即测序技术估计每个样本中每个基因丰度的不确定性。如果相同RNA样本的等分部分被测序,那么特定基因的读取计数应根据泊松定律变化(30)。如果测序变异为泊松,则可以显示(“材料和方法”部分)生物复制库之间每个计数的平方变异系数(CV)分别是技术变异和生物变异的平方变异的总和,
生物变异系数(BCV)是基因(未知)真实丰度在复制RNA样本之间变化的变异系数。它代表了如果测序深度可以无限增加的话,生物复制之间保持的变异系数。技术CV随着计数大小的增加而减小。而BCV则不然。因此,BCV可能是高计数基因不确定性的主要来源,因此可靠的BCV估计对于RNA-Seq实验中差异表达的实际评估至关重要。如果每个基因的丰度在不同的复制RNA样本之间变化,使得基因标准偏差与基因平均值成比例,这是物理量测量的一种常见特性,那么可以合理地假设BCV在不同基因之间近似恒定。然而,我们考虑到BCV可能在基因之间发生变化,也可能在基因表达或预期计数方面表现出系统性趋势。
BCV的大小比真实基因丰度所遵循的确切概率定律更重要。为了数学上的方便,我们假设真实的基因丰度在复制RNA样本之间遵循伽马分布规律。这意味着读取计数遵循负二项式概率定律。
多因素实验的线性模型
线性模型用于描述多因素微阵列实验的方法已经很成熟(18,19)。虽然线性模型与正态分布数据相关,但可以使用GLM分析负二项式计数数据,其方法在所有重要方面都与正态线性模型非常类似。我们假设以解释性协变量表示的预期读取计数为对数线性模型,这些协变量捕获了应用于每个RNA样本的处理条件(“材料和方法”部分)。总库大小N个我充当抵消在线性模型预测器中,捕获计数对序列深度的依赖性。库大小可以定义为映射读取的总数,也可以根据数据进行估计,以影响不同库之间的相对规范化(26).
GLM是非线性模型,必须对每个基因的参数进行迭代估计。提出了一种直观的迭代计算算法来拟合GLM(42),并且几乎所有可用的GLM软件都使用此算法。每个迭代都可以看作是一个最小二乘回归,其中每个计数与总CV成反比2上述定义(43,45)。必须重复模型拟合过程,直到实现收敛。GLM在RNA-Seq数据中的先前应用已经对标准单变量GLM软件进行了基因调用。虽然常用的GLM算法对于单变量数据相当可靠,但不能保证它能够成功收敛,特别是对于非常小或拟合不良的数据集。在RNA-Seq环境中,通常的GLM算法经常失败,对于我们的目的来说不够可靠。我们通过用线搜索修改来改进常用算法来解决这个问题(45)。此修改检查每次迭代的收敛性,减少步长以避免发散。步长反复减半,直到对数似然增加。这确保了算法的收敛性,除非浮点错误介入。线搜索算法在实践中非常可靠。
迭代模型拟合的第二个问题是计算时间。通常的GLM算法需要在每个基因的每次迭代时形成矩阵分解,这是一个巨大的计算负担。为了解决这个问题,我们实现了一种新的、简化的伪纽顿算法,与其他算法相比,它可以更容易地跨基因并行化。在我们的伪纽顿算法中,对模型系数的二阶导数矩阵使用固定近似。首先对线性模型参数化进行变换,使设计矩阵的列正交。然后,二阶导数矩阵由期望信息矩阵近似,如果每个基因的拟合值相等,则会出现期望信息矩阵。这只是单位矩阵的倍数,完全消除了矩阵分解的计算开销。虽然伪纽顿算法平均需要的迭代次数比真纽顿-拉夫森算法或GLM的传统费希尔评分算法略多,但伪纽顿方法与我们的线性搜索策略相结合仍然具有竞争力,并且简化带来的计算收益是巨大的。该算法在R中以这样一种方式实现,即并行地对所有基因进行迭代,而不是一次对一个基因进行迭代。我们的纯R实现在几秒钟内就可以将glm应用于大多数RNA-Seq数据集,而在R中对glm()函数的基因调用通常至少需要几分钟,实际上可能完全由于一个或多个基因的迭代发散而失败。
假设检验
我们的软件允许用户测试线性模型中任何系数的重要性,或线性模型中系数的任何对比度或线性组合的重要性。基因测试是通过计算似然比统计来进行的,以比较系数或对比度等于零的无效假设与不同于零的双边替代假设。在系数或对比度为零的零假设下,对数似然比统计量是渐近卡方分布的。模拟结果表明,似然比检验能够较好地保持其大小,通常能很好地近似于精确检验(23)当后者可用时(未显示数据)。可以使用R中p.adjust函数提供的任何多重测试调整方法。默认情况下,对-通过Benjamini和Hochberg方法调整值以控制错误发现率(47).
生物CV估算
剩下的问题是获得每个基因的BCV的可靠估计值。需要一个近似无偏且在小样本中表现良好的估计量。BCV的最大似然估计会低估BCV,因为需要从相同的数据估计对数线性模型中的系数。我们早期的工作使用精确的条件似然来估计BCV(22,23)。这种方法具有出色的性能,但不容易推广到GLM。相反,我们使用一种近似的条件似然方法,称为APL(48)。APL是惩罚可能性的一种形式。再次,我们以矢量化和计算效率的方式实现了APL计算,而不是逐个基因计算数量。
估算常见分散度
除非有大量生物复制品可用,否则不应考虑单独估计每个基因的BCV。当复制较少时,基因之间的信息共享对于可靠推断至关重要。无论复制的数量如何,适当的信息共享方法都应该带来一些好处。
让克表示基因的平方BCV克,我们称之为分散,分散这个基因的基因。离散度是方差函数中二次项的系数。在基因之间共享信息的最简单方法是假设所有基因共享相同的离散度,因此克 = ϕ (23)。可通过最大化共享似然函数来估计公共离散度
其中APL克是基因的调整配置文件可能性克(“材料和方法”部分)。这种最大化可以通过多种方式在数值上实现,例如通过无导数近似牛顿算法(49).
估计趋势分散
对常见色散的概括是对色散进行建模克作为每个基因的平均读取计数的平滑函数(25)。我们的软件提供了许多方法来实现这一点。一种简单的非参数方法是通过平均读取数将基因划分为各个基因库,估计每个基因库中的常见离散度,然后通过这些二进制离散度拟合黄土或样条曲线。一种更复杂的方法是局部加权APL。在这种方法中克通过进行局部共享对数似然来估计,该似然是基因的APL的加权平均值克以及其相邻基因的平均读计数。
估算基因离散度
在实际科学应用中,单个基因更有可能根据其基因组序列、基因组长度、表达水平或生物功能而具有单个BCV。我们寻求在完全个别的基因分散之间的折衷克通过扩展Robinson和Smyth提出的加权似然经验贝叶斯方法,完全共享值(22)。在这种方法中,克通过最大化估计
哪里G公司0是共享可能性和APL的权重Sg公司(ϕ克)是本地共享的log-likelihood。这种加权似然方法可以用经验贝叶斯术语来解释,共享似然是的先验分布克加权似然作为后验。先验分布可以认为是由对一组G公司0基因。先前基因的数量G公司0因此,表示相对于基因的实际观测数据分配给先验的权重克.最佳选择G公司0取决于基因间BCV的变异性。当基因之间的BCV恒定时,较大的值是最好的。当BCV在基因之间差异很大时,较小的值是最佳的。我们已经发现G公司0 = 20/df在广泛的实际数据集上给出了良好的结果,其中df是估计BCV的剩余自由度。对于多组实验,df是库的数量减去不同治疗组的数量。默认设置意味着,无论库的实际数量如何,优先权都有20个自由度的权重来估计BCVn个或实验设计的复杂性。
经验贝叶斯方法的一个理想结果是,基因分散或多或少地向先验值挤压,这取决于个体基因分散估计的可靠性。计数非常低的基因为估计其自身的离散度提供的统计信息相对较少,因此,在这些情况下,先验值占主导地位,基因离散度严重挤压到总体趋势。
为了计算方便,在可能的色散值网格上评估遗传和共享APL函数。使用三次样条曲线在网格上插值每个基因的APL值,并将样条曲线的最大值作为基因离散估计。计算数万个基因的共同和基因离散度大约需要20 在笔记本电脑上。
口腔鳞状细胞癌
最近的一项研究调查了口腔鳞状细胞癌(OSCC)中的差异基因表达(50)。该研究使用应用生物系统SOLiD系统构建了三名口腔鳞癌患者肿瘤和匹配正常组织的RNA-Seq图谱。最初的分析使用了一种直观但特殊的程序来识别差异表达的基因。基因首先根据肿瘤和正常患者的折叠变化进行排序。在三名患者中,按中位数排序,前300个上调基因和前300个下调基因被选为差异表达基因(50)。这个简单的分析要求一个基因在两个患者中排名较高,然后忽略第三个患者的fold-change。这足以获得有趣的生物学结果,但不允许进行任何统计显著性评估。它还将所有折叠变化视为同等可靠,而不管计数的大小。
在这里,我们描述了一种更为正式的分析,评估与生物变异相关的统计显著性。首先,我们从下载了读取计数数据补充表S1图什的等。(50)。该表给出了由RefSeq转录本汇总的读取计数,并进行筛选,以仅包括所有三名患者中至少一个组织(肿瘤或正常组织)的至少50个对齐读取的转录本。研究的全序列数据可从GEO数据库中获得(网址:www.ncbi.nlm.nih.gov/geo,加入编号GSE20116标准),但根据汇总的计数进行工作可以确保我们的分析基于与原始分析相同的计数。我们使用Bioconductor注释包org.Hs.eg.db(版本2.5.0)将RefSeq标识符映射到最新的官方基因符号,丢弃数据库中不再存在的任何RefSeq标识。选择外显子数量最多的RefSeq转录本来代表每个独特的基因,并删除多余的RefSeg转录本。这只剩下10只 464个转录本,每个转录本代表一个独特的基因。然后使用M值尺度归一化的加权修剪平均值来估计有效库大小(26).
三种正常组织轮廓之间的总(常见)BCV估计为40%(图1)。三种肿瘤组织之间的BCV明显高于52%,表明肿瘤比正常组织更异质。因此,检测至少两类差异表达的基因是有意义的:第一,那些在所有肿瘤中与匹配的正常组织中一致不同的基因,第二,那些表现出特定于三个肿瘤中一个或两个肿瘤的表达变化的基因。
图1。
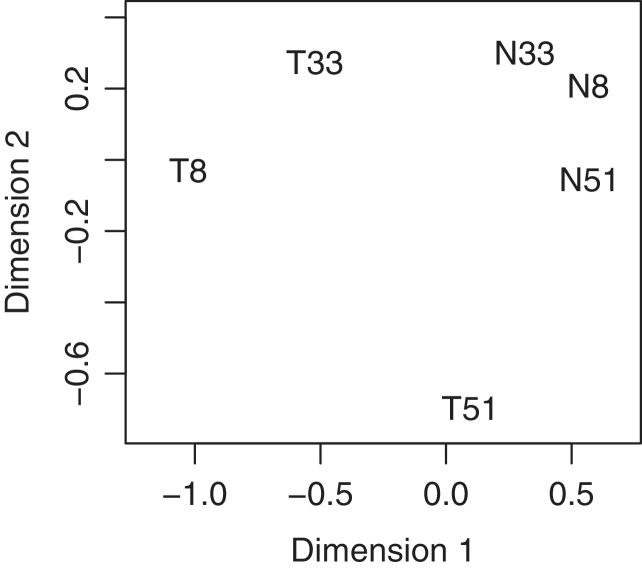
鳞状细胞癌轮廓的多维标度图,其中距离对应于成对样本之间的BCV。根据500个最异质的基因计算成对BCV。样本标有患者编号,肿瘤标记为“T”,正常标记为“N”。第一个绘图维度大致对应于组织来源(正常或肿瘤),第二个绘图维度对应于患者差异。肿瘤样本比正常样本更具异质性。
研究设计有两个解释因素,一个是患者ID,分为三个级别,另一个是组织类型,分为两个级别(正常或肿瘤)。通过将三个连续的对数线性模型拟合到每个基因的读取计数来分析数据(表1)。第一个模型代表了三名患者之间的基线表达差异。第二种是加性模型,允许肿瘤组织与正常组织中一致的相对表达变化。第三种是交互模型,考虑到患者特定的肿瘤效应。
表1。
| 模型 |
解释 |
检测到的基因 |
| 病人 |
基线患者差异 |
|
| 患者+组织 |
一致的肿瘤差异 |
1276 |
| 患者×组织 |
患者特异性肿瘤差异 |
202 |
我们的第一个分析是寻找癌症中与正常组织相比持续差异表达的基因。对于此分析,色散估计基于具有两个剩余自由度的可加模型。所有基因的共同BCV被发现过于简单,39个基因显示出比共同BCV更大的变异性证据(图2)家庭错误率为0.05(51)。允许BCV的丰度趋势并没有减少BCV被拒绝的异常基因的数量(图2)。另一方面,允许基因型BCV,使用先验贝叶斯调节G公司0 = 10,表示没有剩余的不适(图2)。这为在以下分析中使用基因型BCV提供了统计依据。还有一个生物学理由,即三名患者中肿瘤与正常差异不一致的基因将获得更高的BCV估计值,因此在差异表达基因列表中被降级。因此,使用基因型BCV可以让我们关注肿瘤与正常差异一致的基因。
图2。
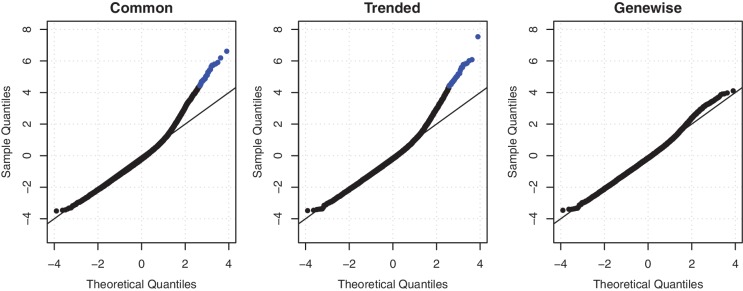
使用普通、趋势或经验贝叶斯基因(分段)离散度的拟合优度统计QQ图。将基因偏差统计转换为正态,并根据理论正态分位数绘制。蓝色的点是那些非常不适合的基因(Holm-adjusted对-值<0.05)。当使用基因分散时,没有基因显示出明显的不匹配。
使用基因BCV值,我们通过比较加法模型和基线模型来测试肿瘤和正常组织之间的差异表达。该分析根据患者之间的基线差异进行调整,其方式类似于计算配对t吨-对每个基因进行测试,但要适应计数数据。以错误发现率(FDR)获得1276个基因 < 0.05 (表1,补充表S1)。在头颈部鳞状细胞癌研究中,这些基因中最显著的是那些先前被确定为肿瘤和正常组织之间差异表达的基因。Yu报道的25个基因中等。(52),18人被列入罗斯福民主共和国的名单 < 0.05 (补充表S2)。另外两个(TNC和FN1)显示折叠变化大于两倍,FDR约为0.4。其余五个基因表现出微小的折叠变化,没有差异表达的证据(补充表S3)。图什等。(50)讨论了9个具有特殊生物学意义的基因。在我们的FDR分析中,确认了其中六个基因(CASQ1、INHBA、MMP1、HMGA2、SHANK2和WIF1)的强差异表达 < 0.001 (补充表S4和S5)。这包括一个通过RT-qPCR验证的基因(HMGA2)。注意,原始研究(50)通过PCR验证了16个基因,但仅鉴定出HMGA2。
为了进一步证明检测到的基因的生物学相关性,我们测试了从MSigDB数据库中富集的精选基因集(53)使用mean-rank基因集富集试验(54)。在FDR < 0.05,这产生了417个富含上调基因的基因集和268个富含下调基因的基因组。显著富集的数据集绝大多数与癌症相关且一致,表明肿瘤中WNT1通路增强,表达特征与其他癌症类似,如基底样乳腺癌(补充表S7和S8)。基因本体分析(55)发现146个GO术语富含上调基因,264个术语富含下调基因。表达上调基因的GO术语往往与细胞发育、增殖和分化以及与肿瘤发展相一致的相关过程有关(补充表S9和S10).
接下来,我们寻找肿瘤异质性与正常差异的基因。理想情况下,该分析应在同一患者的独立组织提取物之间相对于BCV进行。然而,交互模型完全符合可用数据,没有剩余自由度,因此不能用于估计BCV。相反,我们使用根据三名正常患者之间的差异估计的基因BCV进行此分析。这些BCV代表患者间的差异,而非患者内的差异,因此应该高估期望的BCV。因此,我们的分析在一定程度上是保守的对-值和FDR。正常患者之间的BCV在大小上通常与加性模型中的BCV相似,因此保守性可能相对较小。使用这些保守的BCV,对相互作用和加性模型进行比较,得出202个FDR差异表达基因 < 0.05. 在这项分析中排名第一的基因是CDKN2B,它是由Tuch鉴定的等。(50)根据患者8中表达水平与拷贝数变化的相关性,确定其生物学意义。其他两个基因(CCND1,CTTN)同样由Tuch鉴定等。(50)在我们的交互分析中,FDR约为0.1(补充表S6).
模拟研究
通过仿真研究了我们的色散估计器的性能。模拟的第一个场景是最简单的设计,使用一组三个复制库,所有基因的真实BCV恒定值为0.4,以匹配癌症数据。在这个简单的场景中,条件似然提供了偏差最小和最准确的离散估计,Cox–Reid具有很好的可比性(图3a) ●●●●。通常用于基于皮尔逊或偏差残差的广义线性模型的其他估计量表现不佳。这些结果与之前的模拟结果一致(23).
图3。
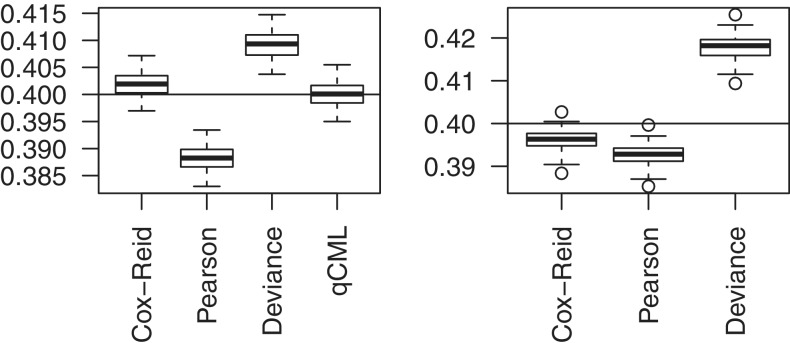
100个模拟数据集的常见BCV估计值的箱线图。左侧面板显示了一组病例的结果,组中有三个重复样本。右侧面板显示两组三块成对设计的结果。水平线表示真正的普通BCV值为0.4,与癌症病例研究相匹配。条件最大似然(qCML)在前一种情况下是最准确的。对于广义线性模型,Cox–Reid APL表现最佳。
第二种情况与癌症病例研究相匹配,有六个文库和一个2 × 3个加性对数线性模型拟合到每个基因。在这种情况下,条件可能性不适用,考克斯-里德APL是其余可能性中表现最好的(图3b) ●●●●。
接下来,我们生成了随机真分散度(BCV2)根据逆chi平方分布,使用相同的2 × 3如前所述的设计。在这种情况下,经验贝叶斯提供了基因离散度的最精确估计。当先前的重量G公司0在10–12范围内,对应于20–24先前自由度(图4)。都不是单独的基因估计(G公司0 = 0)也不常见分散(G公司0 = ∞) 表现也不错。
图4。
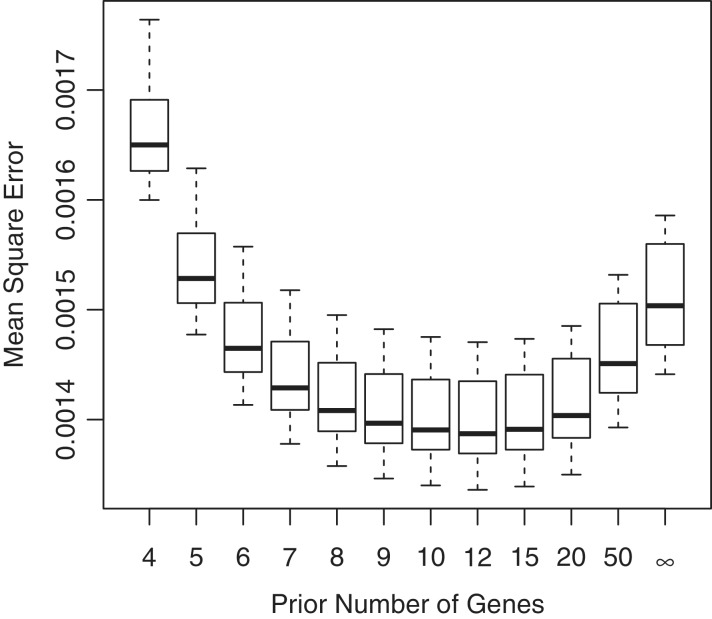
经验贝叶斯基因离散度估计真实离散度(BCV)的均方误差2),当真正的分散是随机产生的。在这种情况下,最优的先验权重是10–12个先验基因,相当于20–24个先验自由度。普通BCV估计量等价于对先验值使用无限权重。方框图显示了10个模拟的结果。
讨论
本文中描述的方法在软件包edgeR中实现(24),作为开源基因组软件的Bioconder项目的一部分提供(56)。这些方法为使用RNA-Seq技术分析基因表达实验中的读取计数提供了一种灵活而强大的方法。基于负二项分布的模型有助于直观地解释生物变异和技术变异的分离。广义线性模型允许任意复杂的实验。经验贝叶斯方法允许基因特异性变异,即使在生物复制相对较少的情况下,这种方法仍然有用。
癌症数据的案例研究分析证明了广义线性模型方法在配对设计中检测差异表达,甚至检测肿瘤特异性表达变化的能力。结果比更特别的方法更详细、更丰富。该案例研究还表明,需要考虑基因特异性变异,而不是假设BCV恒定或丰度-变异关系恒定。除了其他优点外,基因型BCV的估计允许主要分析通过对结果不一致的基因进行去优先化,集中于生物复制之间一致的变化。
生物变异的估计至关重要。例如,基于泊松模型的统计方法会大大低估生物复制数据的可变性,并可能导致大量错误发现。这里开发的管道为将生物变异纳入分析提供了一种可行的方法,即使是在只复制一种条件的情况下进行尽可能小的实验。这并不是要淡化为手头的实验获得科学合理数量的复制品的重要性(27,34)。相反,所提供的方法允许数据分析师充分利用可用的任何数据。
在癌症病例研究中观察到的40%的BCV是我们在其他RNA-Seq或人类受试者的深度SAGE研究中所观察到的典型BCV。另一方面,根据我们的经验,用基因相同的模式生物进行的实验往往在重复之间产生较小的变异性,通常约为BCV的10%(数据未显示)。
我们的数值实现解决了许多与将非线性模型拟合到基因组数据相关的常见问题。我们的模型拟合速度非常快,并且具有可靠的收敛性。对于典型的数据集,在笔记本电脑上拟合基因广义线性模型、假设检验和折迭估计只需一秒钟。基因离散度的经验贝叶斯估计,一个需要许多模型拟合的过程,大约需要20 在笔记本电脑上。仿真表明,Cox–Reid调整后的轮廓似然和经验Bayes估计量能够在复杂情况下返回BCV的准确估计值。当生物变异具有基因特异性时,经验贝叶斯估计可以提供一种有利的折衷方案,优于普通离散或单独的基因离散的两个极端。
在本文中,我们重点关注了基因分析,但该软件也可以用于执行外显子水平的分析,或者实际上用于分析其他基因组特征的读取计数。这篇文章的重点是RNA-Seq和基因表达,但该方法和软件适用于其他类型基因组数据的差异计数分析。这些应用包括使用甲基化DNA免疫沉淀测序(MeDIP-Seq)搜索差异甲基化启动子(57,58),ChIP-Seq用于发现转录因子结合或表观遗传组蛋白标记的差异富集区域,串联质谱中光谱计数的差异分析(59)或宏基因组学研究中的物种计数分析(60)。所有这些技术都产生了基因组规模的计数数据,在此基础上,本文所述的方法可以卓有成效地发挥作用。
除了本文介绍的功能和选项外,edgeR软件还包含许多功能和选项,为RNA-Seq数据分析提供了灵活的可能性。例如,在对数线性模型中使用偏移量可以很容易地适应非线性规范化过程,包括基于分位数规范化或GC-序列内容的程序(61)。广义线性模型的使用还提供了将定量权重合并的潜力,从而将RNA样本的质量权重集成到差异表达分析中,就像微阵列一样(62).
补充数据
补充数据参见NAR Online:补充表1-10。
基金
国家卫生和医学研究委员会(向G.K.S.提供项目拨款490036和研究奖学金);澳大利亚政府(Y.C.获得澳大利亚研究生研究奖)。开放获取费用的资金来源:国家卫生和医学研究委员会项目拨款490036。
利益冲突声明。未声明。
致谢
感谢马克·罗宾逊(Mark Robinson)的许多有益讨论,以及布莱恩·图什(Brian Tuch)对鳞癌研究数据的建议。
参考文献
-
1国家人类基因组研究所(2011年)。DNA测序成本。http://www.genome.gov/序列成本/[谷歌学者]
-
2Morrissy AS、Morin RD、Delaney A、Zeng T、McDonald H、Jones S、Zhao Y、Hirst M、Marra MA。癌症基因表达谱分析的下一代标签测序。基因组研究2009;19:1825–1835. doi:10.1101/gr.094482.109。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
三。‘t Hoen PAC、Ariyurek Y、Thygesen HH、Vreugdenhil E、Vossen RHAM、Menezes RXD、Boer JM、Ommen GJBV、Dunnen JTD。基于深度序列的表达分析表明,在五种微阵列平台上,在稳健性、分辨率和实验室间可移植性方面取得了重大进展。核酸研究2008;第36页:e141。doi:10.1093/nar/gkn705。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
4Wu ZJ、Meyer CA、Choudhury S、Shipitsin M、Maruyama R、Bessarabova M、Nikolskaya T、Sukumar S、Schwartzman A、Liu JS等。使用SAGE-Seq对人类乳腺组织样本进行基因表达谱分析。基因组研究2010;20:1730–1739. doi:10.1101/gr.108217.110。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
5Mortazavi A、Williams BA、Mccue K、Schaeffer L、Wold B.通过RNA-Seq对哺乳动物转录体进行定位和量化。自然法。2008;5:621–628. doi:10.1038/nmeth.1226。[内政部] [公共医学] [谷歌学者]
-
6Cloonan N、Forrest ARR、Kolle G、Gardiner BBA、Faulkner GJ、Brown MK、Taylor DF、Steptoe AL、Wani S、Bethel G等。通过大规模信使核糖核酸测序进行干细胞转录组分析。自然法。2008;5:613–619. doi:10.1038/nmeth.1223。[内政部] [公共医学] [谷歌学者]
-
7Wang Z,Gerstein M,Snyder M.RNA-Seq:转录组学的革命性工具。Nat.Rev.基因。2009年;10:57–63. doi:10.1038/nrg2484。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
8Ozsolak F,Milos PM。RNA测序:进展、挑战和机遇。Nat.Rev.基因。2011;12:87–98。doi:10.1038/nrg2934。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
9Li B,Ruotti V,Stewart RM,Thomson JA,Dewey CN.带读映射不确定性的RNA-Seq基因表达估计。生物信息学。2010;26:493–500. doi:10.1093/bioinformatics/btp692。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
10Tang F、Barbacioru C、Wang Y、Nordman E、Lee C、Xu N、Wang X、Bodeau J、Tuch BB、Siddiqui A等。单细胞mRNA-Seq全转录组分析。自然法。2009年;6:377-82。doi:10.1038/nmeth.1315。[内政部] [公共医学] [谷歌学者]
-
11Sultan M、Schulz MH、Richard H、Magen A、Klingenhoff A、Scherf M、Seifert M、Borodina T、Soldatov A、Parkhomchuk D等。通过人类转录组的深度测序对基因活性和选择性剪接的全球观点。科学。2008;321:956–960. doi:10.1126/science.1160342。[内政部] [公共医学] [谷歌学者]
-
12Pan Q,Shai O,Lee LJ,Frey BJ,Blencowe BJ。通过高通量测序深入研究人类转录组中的选择性剪接复杂性。自然遗传学。2008;40:1413–1415. doi:10.1038/ng.259。[内政部] [公共医学] [谷歌学者]
-
13Denoeud F、Aury JM、Silva CD、Noel B、Rogier O、Delledonne M、Morgante M、Valle G、Wincker P、Scarpelli C等。用大规模RNA测序注释基因组。基因组生物学。2008;9:R175。doi:10.1186/gb-2008-9-12-r175。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
14李杰、姜浩、王浩。RNA-Seq数据中短读速率的非均匀性建模。基因组生物学。2010;11:R50。doi:10.1186/gb-2010-11-5-r50。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
15Trannell C、Williams BA、Pertea G、Mortazavi A、Kwan G、van Baren MJ、Salzberg SL、Wold BJ、Pachter L.通过RNA-Seq对转录物进行组装和量化,揭示了细胞分化过程中未标记的转录物和亚型转换。自然生物技术。2010;28:511–5. doi:10.1038/nbt.1621。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
16Degner JF、Marioni JC、Pai AA、Pickrell JK、Nkadori E、Gilad Y、Pritchard JK。读图偏差对从RNA测序数据中检测等位基因特异性表达的影响。生物信息学。2009年;25:3207–3212. doi:10.1093/bioinformatics/btp579。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
17Montgomery SB、Sammeth M、Gutierrez-Arcelus M、Lach RP、Ingle C、Nisbett J、Guigo R、Dermitzakis ET。在高加索人群中使用第二代测序的转录组遗传学。自然。2010;464:773–777. doi:10.1038/nature08903。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
18Wright G,Simon R.小型微阵列实验中检测差异基因表达的随机方差模型。生物信息学。2003年;19:2448–2455. doi:10.1093/bioinformatics/btg345。[内政部] [公共医学] [谷歌学者]
-
19用于评估微阵列实验中差异表达的线性模型和经验贝叶斯方法。统计应用程序。遗传学。摩尔。《生物》2004;3网址:10.2202/1544-6115.1027。第3条。[内政部] [公共医学] [谷歌学者]
-
20Oshlack A,Robinson MD,Young MD。从RNA-seq读取到差异表达结果。基因组生物学。2010;11:220. doi:10.1186/gb-2010-11-12-220。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
21Langmead B、Hansen KD、Leek JT。用Myrna进行云级RNA测序差异表达分析。基因组生物学。2010;11:R83。doi:10.1186/gb-2010-11-8-r83。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
22Robinson医学博士、Smyth GK。用于评估标记丰度差异的适度统计测试。生物信息学。2007年;23:2881–2887. doi:10.1093/bioinformatics/btm453。[内政部] [公共医学] [谷歌学者]
-
23Robinson医学博士、Smyth GK。负二项离散度的小样本估计及其在SAGE数据中的应用。生物统计学。2008;9:321–332。doi:10.1093/biostatistics/kxm030。[内政部] [公共医学] [谷歌学者]
-
24Robinson医学博士、McCarthy DJ、Smyth GK。edgeR:用于数字基因表达数据差异表达分析的生物导体包。生物信息学。2010;26:139–40. doi:10.1093/bioinformatics/btp616。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
25Anders S,Huber W.序列计数数据的差异表达分析。基因组生物学。2010;11:R106。doi:10.1186/gb-2010-11-10-r106。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
26Robinson MD,Oshlack A.RNA-seq数据差异表达分析的标度归一化方法。基因组生物学。2010;11:R25。doi:10.1186/gb-2010-11-3-r25。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
27Hansen KD、Wu Z、Irizarry RA、Leek JT。测序技术并不能消除生物变异性。国家生物技术。2011;29:572–573. doi:10.1038/nbt.1910。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
28Tusher VG,Tibshirani R,Chu G.应用于电离辐射反应的微阵列显著性分析。程序。美国国家科学院。科学。美国2001年;第98:5116页。doi:10.1073/pnas.091062498。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
29Baldi P,Long A.微阵列表达数据分析的贝叶斯框架:基因变化的正则化t检验和统计推断。生物信息学。2001;17:509. doi:10.1093/bioinformatics/17.6.509。[内政部] [公共医学] [谷歌学者]
-
30Marioni JC、Mason CE、Mane SM、Stephens M、Gilad Y.RNA-seq:技术再现性评估和与基因表达阵列的比较。基因组研究2008;18:1509–1517. doi:10.1101/gr.079558.108。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
31Baggerly K,Deng L,Morris J,Aldaz C.SAGE的过度分散逻辑回归:建模多组和协变量。BMC生物信息学。2004;5:144. doi:10.1186/1471-2105-5-144。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
32Lu J,Tomfohr J,Kepler T。识别多个SAGE库中的差异表达:过度分散的对数线性模型方法。BMC生物信息学。2005;6:165. doi:10.1186/1471-2105-6-165。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
33Blekhman R、Marioni JC、Zumbo P、Stephens M、Gilad Y.灵长类动物的性别特异性和谱系特异性选择性剪接。基因组研究2010;20:180–189. doi:10.10101/gr.099226.109。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
34Auer PL,Doerge RW。RNA测序数据的统计设计和分析。遗传学。2010;185:405–416. doi:10.1534/genetics.110.114983。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
35Srivastava S,Chen L.改进RNA-seq数据分析的双参数广义泊松模型。核酸研究2010;38:e170。doi:10.1093/nar/gkq670。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
36Auer PL,Doerge RW。用于测试RNA-Seq数据的两阶段泊松模型。统计应用程序。遗传学。摩尔。生物杂志2011;10:1–28.[谷歌学者]
-
37周义赫,夏克,赖特。一种强大而灵活的RNA序列计数数据分析方法。生物信息学。2011;27:2672–2678. doi:10.1093/bioinformatics/btr449。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
38Vencio RZ,Brentani H,Pereira CA。在SAGE分析中使用可信度区间代替假设检验。生物信息学。2003年;19:2461–2464. doi:10.1093/bioinformatics/btg357。[内政部] [公共医学] [谷歌学者]
-
39Víncio RZN、Brentani H、Patráo DFC、Pereira CAB。基因表达序列分析(SAGE)BMC生物信息学中类内生物变异性的贝叶斯模型。2004;5:119. doi:10.1186/1471-2105-5-119。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
40Hardcastle TJ,Kelly KA公司。baySeq:识别序列计数数据中差异表达的经验贝叶斯方法。BMC生物信息学。2010;11:422. doi:10.1186/1471-2105-11-422。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
41Bullard J,Purdom E,Hansen K,Dudoit S。mRNA-Seq实验中归一化和差异表达统计方法的评估。BMC生物信息学。2010;18:11至94。doi:10.186/1471-2105-11-94。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
42Nelder JA,Wedderburn RWM公司。广义线性模型。J.罗伊。Stat.Soc.A.1972;135:370–384。[谷歌学者]
-
43McCullagh P,日本内尔德。广义线性模型。第2版。佛罗里达州博卡拉顿:查普曼和霍尔/CRC;1989[谷歌学者]
-
44Osborne MR.Fisher的评分方法。国际统计评论。1992;60:99–117.[谷歌学者]
-
45Smyth GK公司。优化和非线性方程。收件人:Armitage P,Colton T,编辑。生物统计学百科全书。伦敦:威利;1998年,第3174–3180页。[谷歌学者]
-
46Stewart G.计算机科学和应用数学。纽约:学术出版社;1973.矩阵计算导论。[谷歌学者]
-
47Benjamini Y,Hochberg Y。控制错误发现率:一种实用而强大的多重测试方法。J.罗伊。1995年国家社会委员会;57:289–300.[谷歌学者]
-
48Cox DR,Reid N.参数正交性和近似条件推理。J.罗伊。1987年国家社会委员会;49:1–39.[谷歌学者]
-
49无导数最小化的Brent R.算法。新泽西州恩格尔伍德克利夫斯:普伦蒂斯·霍尔;1973[谷歌学者]
-
50Tuch BB、Laborde RR、Xu X、Gu J、Chung CB、Monighetti CK、Stanley SJ、Olsen KD、Kasperbauer JL、Moore EJ等。肿瘤转录组测序揭示了与拷贝数改变相关的等位基因表达失衡。《公共科学图书馆·综合》。2010;5:e9317。doi:10.1371/journal.pone.0009317。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
51Holm S.一种简单的顺序拒绝多重测试程序。扫描。J.Stat.1979;6:65–70.[谷歌学者]
-
52Yu YH,Kuo HK,Chang KW,Rutherford S.头颈部鳞状细胞癌的进化转录组:系统综述。《公共科学图书馆·综合》。2008;3:e3215。doi:10.1371/journal.pone.0003215。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
53Subramanian A、Tamayo P、Mootha VK、Mukherjee S、Ebert BL、Gillette MA、Paulovich A、Pomeroy SL、Golub TR、Lander ES等。基因集富集分析:解释全基因组表达谱的基于知识的方法。程序。美国国家科学院。科学。美国2005年;102:15545-50。doi:10.1073/pnas.0506580102。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
54Michaud J、Simpson KM、Escher R、Buchet-Poyau K、Beissbarth T、Carmichael C、Ritchie ME、Schütz F、Cannon P、Liu M等。RUNX1下游通路和靶基因的综合分析。BMC基因组学。2008;9:363. doi:10.1186/1471-2164-9-363。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
55Young MD、Wakefield MJ、Smyth GK、Oshlack A.RNA-seq的基因本体分析:选择偏差的解释。基因组生物学。2010;11:R14。doi:10.1186/gb-2010-11-2-r14。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
56Gentleman RC、Carey VJ、Bates DM、Bolstad B、Dettling M、Dudoit S、Ellis B、Gautier L、Ge Y、Gentry J等人。生物导体:计算生物学和生物信息学的开放软件开发。基因组生物学。2004;5:R80。doi:10.1186/gb-2004-5-10-r80。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
57Bock C、Tomazou EM、Brinkman A、Müller F、Simmer F、Gu H、Jäger N、Gnirke A、Stunnenberg HG、Meissner A。DNA甲基化的基因组图谱:定量技术比较。自然生物技术。2010;28:1106. doi:10.1038/nbt.1681。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
58Robinson MD、Stirzaker C、Statham AL、Coolen MW、Song JZ、Nair SS、Strbenac D、Speed TP、Clark SJ。基于亲和力的全基因组dna甲基化数据评估:CpG密度、扩增偏差和拷贝数变化的影响。基因组研究2010;20:1719–1729. doi:10.1101/gr.110601.110。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
59Carvalho P,Hewel J,Barbosa V,Yates JR.,第3版通过光谱计数和特征选择确定蛋白质表达水平的差异。遗传学与分子研究2008;7:342–356. doi:10.4238/vol7-2gmr426。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
60White JR,Nagarajan N,Pop M.检测临床宏基因组样本中差异丰富特征的统计方法。公共科学图书馆计算生物学。2009年;5:e1000352。doi:10.1371/journal.pcbi.1000352。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
61Hansen KD,Irizarry RA,Wu Z.使用条件分位数归一化消除RNA-seq数据中的技术变异性。生物统计学。2012;13:204–216. doi:10.1093/biostatistics/kxr054。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
62Ritchie M、Diyagama D、Neilson J、Laar RV、Dobrovic A、Holloway A、Smyth G。微阵列数据分析中的经验阵列质量权重。BMC生物信息学。2006;7点261分。doi:10.1186/1471-2105-7-261。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。