摘要
动机:物种系统发育的估计需要多个基因座,因为不同的基因座可能由于谱系分类不完全而具有不同的树,这是由多物种合并模型建模的。我们最近开发了一种基于聚合的方法ASTRAL,该方法在多物种聚合模型下具有统计一致性,并且在我们检查的数据集上比其他基于聚合的算法更准确。ASTRAL通过使用一组允许的“双分区”约束搜索空间,在多项式时间内运行。尽管对允许的双分区有限制,ASTRAL在统计上是一致的。
结果:我们提出了一个新版本的ASTRAL,我们称之为ASTRAL-II。我们表明,ASTRAL-II比ASTRAL具有实质性优势:它更快,可以分析更大的数据集(多达1000个物种和1000个基因),并且在某些条件下具有更好的准确性。ASTRAL的运行时间为,ASTRAL-II的运行时间为,其中n个是物种数量,k个是基因座的数量X(X)是搜索空间允许的双分区集。
可用性和实施:ASTRAL-II有开源版本,网址为https://github.com/smirarab/ASTRAL网站和使用的数据集位于网址:http://www.cs.utexas.edu/~phylo/datasets/astral2/.
联系人: smirarab@gmail.com
补充信息: 补充的数据可在生物信息学在线。
1引言
由于基因组的不同部分具有不同的进化历史,物种树的估计变得复杂;因此,在不同基因座上获得的不同基因树往往相互冲突,并与真实物种树相冲突。由于不完全谱系排序(ILS)导致的基因树不一致是物种树估计的主要挑战(Degnan和Rosenberg,2009年;爱德华兹,2009;麦迪逊,1997)这是快速辐射的一个特殊问题(在相对较短的时间内发生了几个物种形成事件)。
由于基因树冲突的可能性,物种树估计越来越多地基于多个位点。估计物种树的一种方法是简单地将不同位点的序列比对连接在一起,并在连接比对上估计树。然而,在多物种联合的情况下,基于级联的分析可能在统计上不一致(Roch and Steel,2014年)并可能导致高支撑的错误树(Kubatko和Degnan,2007年). 由于串联分析有可能产生错误的物种树,因此开发了许多方法来解决因ILS导致的基因树不一致。其中一些方法已经过验证统计上一致在多物种融合下(Rannala和Yang,2003年)这意味着,如果有足够多的真基因树,他们将以很高的概率返回真物种树。其中一些基于聚合的方法[例如MP-EST线路接口单元等。(2010)和NJst刘和余(2011)]目前正在广泛使用。
尽管可以使用基于合并的方法,但许多生物数据集对于可用的方法来说太大了;例如,由于计算原因,MP-EST不能用于大型数据集(拜齐德等。, 2014). 其他基于聚合的方法更加有限;例如,*BEAST(Heled和Drummond,2010年),一种共同估计基因树和物种树的方法,不能用于超过25个物种(齐默尔曼等。, 2014). 实际数据集分析中的计算挑战要求开发基于合并的方法来分析更大的数据集;例如,MP-EST无法分析1KP(威克特等。, 2014)由于数据集的大小等问题,大约有100个物种和600个基因的数据集。
ASTRAL公司(米拉拉布等。,2014年a)是为了对这些较大的数据集进行基于合并的分析而开发的。ASTRAL通过将允许的搜索空间限制为那些从输入集导出其双分区的物种树来解决可能的NP-hard问题X(X),由用户提供。在ASTRAL的默认设置中,我们设置X(X)是输入基因树中的所有双分区。使用此设置,ASTRAL在多物种合并下在统计上是一致的X(X)并在多项式时间内运行。ASTRAL对我们在米拉布等。(2014年a); 然而,所有这些数据集都相对较小(最多37种)。我们随后对ASTRAL的评估(我们在这里报告)表明,ASTRAL对于大型数据集和该设置的运行时间快速增加X(X)输入基因树中的双分割会降低ASTRAL在特定模型条件下估计物种树的准确性。特别是,此设置用于X(X)是存在大量分类群、少数基因树或高度不一致的问题。
我们引入了ASTRAL-II,这是ASTRAL的一个新版本n个(其中n个是物种数量),我们将演示如何定义集合X(X)这样ASTRAL就更加健壮,并且可以探索更大的搜索空间。我们还修改了ASTRAL,以便它可以处理输入树中的多段。我们比较了ASTRAL和基于合并的物种树估计方法,以及在一组模拟数据集和生物数据集上使用最大似然(CA-ML)进行连接的方法。我们表明,ASTRAL优于基于合并的方法,提供了改进的准确性,并且能够分析非常大的数据集。特别是,我们显示ASTRAL可以使用单个处理器在大约一天内分析1000个物种和1000个基因。ASTRAL和CA-ML之间的比较表明,只要ILS水平足够高,ASTRAL就更准确,并且在ILS水平很低的情况下接近CA-ML。我们的大量模拟表明,选择最佳方法通常取决于基因树错误的数量、基因数量和不一致程度。在生物数据方面,我们表明,CA-ML和MP-EST之间的一些差异以前归因于这样一个事实,即MP-EST对ILS的解释必须谨慎,因为在ILS下也是一致的ASTRAL-II恢复了类似于CA-ML的拓扑结构。
2背景:ASTRAL-I
给定一组属于k个二进制输入基因树n个分类群,有多组由输入诱导的四叉树。我们将给定树的加权四叉树(WQ)分数定义为给定树也诱导的来自该多集的四叉树的数量。ASTRAL解决的优化问题是找到使WQ得分最大化的物种树(米拉拉布等。,2014年a)。
ASTRAL使用动态编程算法解决了这个问题。每个内部节点未生根的树将叶子集分为三部分,定义为三部分(补充图S14)反之亦然,这样的三分区定义了无根树中的节点。我们使用表示三分割,并可互换地使用三分割和内部节点。每个三分区还定义了一些四元拓扑,这些拓扑将由包含该三分区作为节点的任何树诱导(补充图S14)。米拉拉布等。(2014年a)提出了一个计算两个给定三分区之间共享诱导四叉树数的公式。让和是两个三部分,让对于.共享的四重奏数量T型和如下所示:哪里G公司三给出了{1,2,3}和F类由以下人员提供:米拉拉布等。(2014年a)定义了三部分的总分:哪里是中的内部节点集g、。米拉拉布等。(2014年a)表明物种树的WQ得分可以通过求和计算w个(T型)计算树中的所有节点,然后将总和除以2(每个基因树四分位将被计算两次)。 使用w个函数允许我们使用动态编程来最大化WQ得分。动态编程从集合开始L(左)并递归地将其划分为较小的子集,每次都会找到使得分最大化的除法。为了准确地解决这个问题,需要考虑划分子集的所有方法(这是指数)。然而,我们可以通过定义集合来限制搜索空间X(X)分类群的二分法,并将我们的搜索限制为源于十、。让(因此,是中所有双分区的两个部分的集合X(X)). 在约束搜索中,只有当两部分都出现在因此,动态编程中的递归变成:哪里V(V)(A类)给出上的最优子树的分数A类、和w个定义见方程式(2). 请注意,我们在构建的物种树中允许的双分区仅限于在集合中找到的那些分区十、。ASTRAL-I默认设置X(X)输入基因树中观察到的双分割集。米拉拉布等。(2014年a)证明了使用这个集合X(X)确保ASTRAL的统计一致性。然而,证明仅依赖于以下事实:X(X)包含输入基因树中的所有双分区,因此任何集合X(X)包含这些双分区(也许还有其他分区)也保持了统计一致性。因此, 定理1
让X(X)0表示在输入基因树中发现的双分割集。如果,然后对集合使用ASTRALX(X)在多物种合并模型下具有统计一致性。
3 ASTRAL-II标准
ASTRAL-II有三个新功能:(i)它使用更快的算法进行计算w个,(ii)它通过使用启发式扩展集合X来搜索更大的空间,以及(iii)它可以处理输入中的多段。
3.1运行时间改进
比分w个(方程式2)需要为每个三部分进行计算,并且这种三分制需要评分。ASTRAL-I计算w个在里面每个三分区的时间,但在ASTRAL-II中,我们使用了一个更好的算法,该算法只使用O(运行)(国家银行)时间。在ASTRAL-I中,我们求和O(运行)(国家银行)输入基因树节点,对于每个节点,我们首先计算C类然后计算气使用方程式(1). 我们将分类群的子集表示为位集,这导致O(运行)(n个)计算运行时间C类; 因此,计算每个w个要求在ASTRAL-II中,我们不查看输入基因树中的三部分,而是对所有基因树(任意生根)进行后序遍历,并使用算法1中显示的算法计算得分。
算法1。权重计算:输入是基因树克和三分法.每个部件(例如。X(X))是由物种索引的位集(因此,如果叶为1u个在中X(X)否则为0)。气定义如等式。1。对所有并将结果相加以计算w个定义见方程式(2)。 功能重量() 和空堆栈
对于做
如果u个是一片叶子然后
其他的
从中拉出秒
从中拉出秒
推动(x个,年,z(z))至秒
算法2。计算相似矩阵:叶计数给出了节点下的叶数,很容易进行预计算。
功能获得相似性()
对于和做
对于做
对于做
算法3。使用贪婪共识添加到X:请参阅中函数的详细描述补充表S3.返回贪婪的共识,仅包括具有频率的分支; 如果b条是的,共识中的多面体是随机解决的。更新X(t吨)从树中添加二分t吨到集合X(X); 边进入时t吨用频率标签标记(例如贪婪共识中的频率),则返回任何新的添加到的双分区十、集群(第页)返回由未根节点定义的分类单元分区p.upgma公司(秒,C类)使用相似矩阵运行UPGMA算法秒; 什么时候C类给定,UPGMA由中定义的组启动C.兰德样品(第页)从节点周围的每个子树中选择一个随机分类单元第页和决定(第页,第页)解决多胞胎第页根据一棵树第页在这样的抽样中。操作员将树或矩阵限制为子集。梳状物(O(运行))返回基于的梳状树O(运行),分类群的有序列表。排序依据根据与给定分类单元的相似性降低对分类单元列表进行排序。常量:; MIT=10;后轮驱动=2和. 功能ADDBYGREEDY公司()
对于做
对于做
和
虽然c(c)<最大值做
如果然后
如果然后
对于做
对于每个遍历节点,此算法u个,计算下的分类数u个这是与被计分的三部分中的每一方共享的。这是使用O(运行)(1) 对已经为的子级计算的相同数量求和的计算美国。在叶子上,我们只需要找到三分法的哪一侧包含该叶子,这也可以在中完成O(运行)(1) 使用位集。因此,我们很容易计算C类中的矩阵O(运行)(1) 因此,计算w个对于每一个三分法要求O(运行)(国家银行)运行时间。因此,
定理2
ASTRAL-II的运行时间为.
3.2 X的添加
我们使用以下启发式策略将双分区添加到集合X中。
3.2.1相似矩阵
我们将一对分类群之间的相似性定义为基因树诱导的四元树数量,其中这对分类群出现在四元树的同一侧。我们通过遍历输入基因树的所有节点,任意生根来计算相似度矩阵(算法2)。对于每个节点u个,我们看一看从美国。对于每一对,我们添加相似度得分,其中年是树叶的数量外部下面的子树美国。这将处理每个输入中的每对节点k个基因正好一次,因此需要计算。最终得分可以通过包含任意对的输入四元树的数量进行标准化(算法2中未显示)。我们使用相似度矩阵来计算UPGMA树,并将它的所有双分区添加到X。该矩阵也将用于我们的下一个启发式。
3.2.2贪婪
一组树的贪婪一致性是通过从一棵星型树开始,如果与之前的两分法不冲突,则从输入树中按频率降序添加两分法来获得的。当没有剩余的双分区的频率超过给定阈值或树完全解析时,此过程结束。我们估计了具有不同阈值的基因树的贪婪一致性(算法3)。对于每个共识树中的每个多边形,我们以多种方式解析该多边形,并将这些解析所隐含的两分法添加到集合X中。首先,我们从多边形定义的簇开始,通过将UPGMA应用于相似矩阵来解析该多边形。然后,我们从多胞体的每一侧随机抽取一个分类单元,并使用限制于该子样本的基因树的贪婪一致性来找到多胞体(随机解析剩余的多胞体)的解析。我们至少重复这个过程10次,但如果子样本贪婪一致性树包含足够频繁的新双分区(),我们进行更多的随机采样(我们将迭代次数增加两次)。对于多边形周围的每个随机子样本,我们还通过计算子样本相似矩阵上的UPGMA树来解决它。最后,对于两个第一贪婪阈值和前10个随机子样本来说,我们还使用了第三种策略,可以潜在地添加更多的双分区:对于每个子样本分类单元x个,我们通过根据剩余分类群与的相似性对其进行排序,将多胞体分解为梳状树x个(按降序)。
3.2.3基因树多组
当基因树包含多胞体时,我们还向集合X添加了新的两分法。我们首先计算阈值为0的输入基因树的贪婪一致性,如果贪婪一致性有多胞体,则使用UPGMA进行求解;我们重复这一过程两次,以解释贪婪共识估计中的随机断线。然后,对于每个基因树多胞体,我们使用两个解析的贪婪共识树来推断多胞体的分辨率,并将隐含的分辨率添加到集合X中。
3.3多分支输入基因树
虽然假设真基因树是完全解析的(二进制),但在某些情况下,估计的ML树会产生多组。当输入的每个节点定义的不是三分法而是分类群集合的多分法时,将ASTRAL扩展到包含多组的输入需要解决WQ树问题。我们从一个基本观察开始:每个断然的基因树诱导的四叉树映射到基因树中的两个节点不管基因树是否是二元的(补充图S14). 换句话说,只映射到基因树的一个节点的诱导四元树是未解决。当最大化四元树支持时,这些未解决的基因树四元树无关紧要,需要忽略。现在,考虑一个程度的多元论d日,它将分类群划分为d日部分。围绕多胞体的三个部分的任何选择都定义了一个三分割。从这个三分法的一部分中选择两个分类单元,从其余两部分中选择一个分类单元都会产生一个解析的四分树,每个解析的四元树正好映射到我们的多分树中的两个节点。因此,ASTRAL的所有算法假设都保持不变,只要每个度d日节点,我们将其视为三部分。因此,对于三分法和一个多分区,我们让为所有人和我们推广了方程(1)收件人:哪里P(P)三是大小为3的所有有序子集的集合.扩展算法1以计算方程式(三)简单明了。 在存在多胞体的情况下,运行时间分析可能会发生变化,因为分析每一个多胞体都需要时间立方的度数,度数可以随着n.(名词)。不难看出,最坏的情况是所有基因树都有一个多角体; 在这种情况下,运行时间为.
3.4统计一致性
ASTRAL-II在多物种合并模型下具有统计一致性。
为开发ASTRAL-II而对ASTRAL-I进行的更改影响了运行时间,扩大了搜索空间,并允许它使用多组分分析基因树。在多物种合并模型下,所有基因树都是二元的。如定理1所示,只要集合X(X)包含输入基因树中的所有双分区,ASTRAL在统计上是一致的。定理如下。
4实验装置
4.1模拟程序
我们使用SimPhy(https://github.com/adamallo/SimPhy)模拟物种树和基因树(以突变单位产生),然后使用“不可磨灭”(Fletcher和Yang,2009年)用不同的长度和模型参数模拟基因树中的核苷酸序列。我们根据这些模拟的基因比对来估计基因树,然后将其用于基于聚合的分析。
我们模拟了11个模型条件,将其分为两个数据集,其中一个模型条件出现在两个数据集中。我们使用SimPhy根据Yule过程模拟物种树,以分类群数量、最大树长和物种形成率为特征(此组合定义了模型条件)。在六种模型条件下(形成数据集I),我们将分类群数量固定为200个,并改变树长(500K、2M和10M代)和物种形成率(每代1e-6和1e-7)。树木长度影响ILS的数量,长度越短,树枝越短,ILS水平越高(补充图1a). 物种形成率影响物种形成事件是发生在靠近顶端(1e-06)还是靠近底部(1e-07)。不同的树形(即树长和物种形成率的组合)从相对较低的水平开始产生不同水平的ILS(真实基因树和物种树之间大约10%的距离,用Robinson–Foulds(RF)距离测量);罗宾逊与犯规(1981)]并上升到非常高(约70%RF)。在其余的模型条件下(形成数据集II),我们将树的形状固定为2 M/1e-06,并将分类群的数量设置为10、50、100、200、500和1000。因此,这两个数据集中都出现了200个分类群和2 M/1e-6树形的模型条件。
对于每个模型条件,我们模拟了50种树木,形成50个重复。在每棵物种树上,根据多物种联合模型模拟了1000棵基因树,种群规模固定为200000(脊椎动物的合理值)。SimPhy使用各种速率参数和速率异质性修改器将基因树分支长度转换为突变单位,引入了与分子钟的偏差和基因之间的速率异质性(参见补充表S1参数;模拟脚本可在http://www.cs.utexas.edu/users/phylo/software/astral/)。
我们使用Indelible和GTR+Γ模型模拟了非依赖性基因比对。首先,对于每个重复,从中统一得出两个参数μ和σ和分别是。然后,从具有μ和σ参数的对数正态分布中提取该复制中每个基因的序列长度(因此,平均序列长度均匀分布在300bp和1500bp之间)。GTR+Γ参数是从Dirichlet分布中得出的,该分布的参数是使用ML从真实的生物数据集集合中估计的(详细信息见补充材料)。
4.2基因树估计
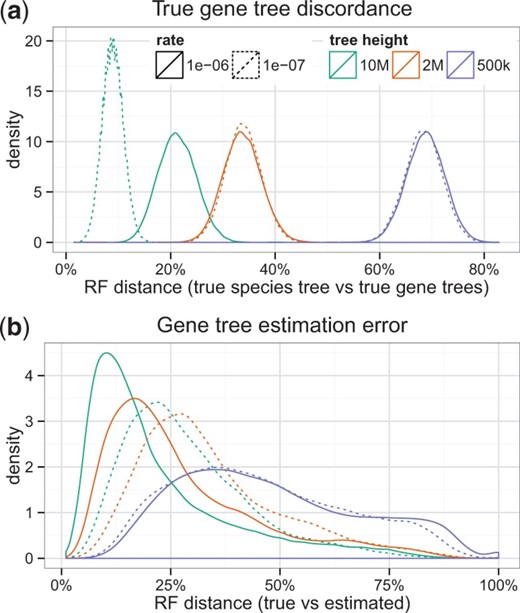
以前的研究(线路接口单元等。, 2011)已经证明了FastTree-II(价格等。, 2010)通常,在估计树拓扑时与更广泛的ML启发式(如RAxML)一样准确(Stamatakis 2014年),同时速度更快。在我们的模拟研究中,我们使用FastTree估计了从10到1000个物种的55万个基因树。图1b显示了基因树估计误差的分布,并证明我们模拟了广泛水平的基因树误差。树形参数对树误差有影响;更多的ILS和更深的物种形成导致更高水平的基因树错误。此外,平均基因树估计误差在不同重复之间存在差异,每个重复中1000个基因的基因树误差差异很大;分类群的数量对基因树估计误差的影响很小(补充图S1)。
图1。
模拟的特征。(一)数据集I的真实物种树和真实基因树(1000个基因的50个重复)之间的RF距离。树高直接影响真实不一致的数量;物种形成率仅影响10M树长的真基因树不一致性。分类群的数量对不一致性有适度的影响(参见补充图S13). (b条)数据集I的真基因树和估计基因树之间的RF距离。另请参阅补充图S1重复基因树间和重复基因树内的错误分布
当序列比对无法区分竞争树分辨率时,FastTree可以输出多段。我们删除了所有50%以上内部节点为多胞体的基因树。这种修剪为200个分类单元/500 K/1e-06和50个分类单元模型条件的三个重复、100个分类单元模式条件的两个重复和10个分类单元模块条件的一个重复留下了不到500个基因。这9个重复(550个)从我们的分析中删除。
4.3物种树方法
我们运行所有方法的最大运行时间为4天,内存为24GB。我们使用FastTree将ASTRAL-I与ASTRAL-II进行比较,并将ASTRAL-II与NJst和CA-ML进行比较。MP-EST仅在时间限制内完成最多100个分类群的数据集。由于它的运行时间,我们为每个分析运行一次MP-EST(一个随机种子数)。NJst、ASTRAL-I和MP-EST不能处理多染色体;因此,我们在这些方法的输入中随机分解了多段。我们还用随机解析的多组分在基因树上运行ASTRAL-II,并观察到与用多组分基因树运行ASTRAL-II没有差异(补充图S12). 因此,ASTRAL-II和其他方法之间的差异并不是由于多面体的随机分辨率造成的。
4.4评价标准
我们根据物种树误差评估方法,并评估基于聚合的方法的运行时间。使用标准射频距离测量物种树误差。在异构condor集群上测量了摘要方法的运行时间,并给出了挂钟的运行时间。
5仿真结果
我们首先比较ASTRAL-II和ASTRAL-I的精度和运行时间(RQ1)。接下来,我们将关注ASTRAL-II,并将其与其他基于聚合的方法(RQ2)进行比较,然后将其与CA-ML(RQ3)进行比较。这个问题使我们更深入地分析了基因树估计误差对各种方法(RQ4)准确性的影响。最后,我们评估了输入基因树中折叠低支持度分支对ASTRAL-II(RQ5)准确性的影响。
5.1 RQ1:ASTRAL-I与ASTRAL-II
5.1.1搜索空间
ASTRAL-II为搜索空间添加了额外的双分区,使其能够探索更大的搜索空间;这有助于提高ASTRAL-II相对于ASTRAL-I的精度。在我们的模拟中,改进的程度取决于模型条件(表1). 在数据集I中,ILS水平最低或ILS水平中等且最近物种形成,ASTRAL-I和ASTRAL-II的误差都极低(补充图S2)添加额外的双分区并没有发现实质性的改进(表1). 在2M长度和深种的情况下,ASTRAL-II显著改善了ASTRAL-I,改善范围从1000个基因的3.5%到50个基因的10.1%。在高ILS条件下观察到了最显著的差异,ASTRAL-I的表现极差(补充图S2),但ASTRAL-II将误差减少了约40%(表1). 数据集II的结果表明,添加额外的双分割的效果也取决于预期的分类群数量(表1). 有了这种固定的树形,ASTRAL-I对于多达200个分类群的准确度与ASTRAL-II一样,但对于500个或更多分类群,ASTRAL-II具有很大的优势(高达9%)。正如预期的那样,ASTRAL-II的优势在基因较少的情况下更大,而在基因较多的情况下则减弱。
表1。与ASTRAL-I相比,ASTRAL-II获得的物种树误差减少
| 数据集I[200分类群,不同树形(列)和基因数(行)]. |
|---|
| . | 10e-6(最近). | 10e-7(深). |
|---|
| 1000万. | 200万. | 500公里. | 1000万. | 200万. | 500公里. |
|---|
| 50 | 0.2 ± 0.2 | 0.7±0.3 | 37.9 ± 1.0 | 1.7 ± 0.6 | 10.1 ± 0.9 | 38.7 ± 0.9 |
| 200 | 0.0 ± 0.1 | 0.2 ± 0.1 | 41.0±1.1 | 0.7 ± 0.3 | 7.4 ± 0.7 | 41.4 ± 1.0 |
| 1000 | 0.0 ± 0.0 | 0.2 ± 0.1 | 39.2 ± 1.2 | 0.0 ± 0.0 | 3.5 ± 0.7 | 41.4 ± 1.1 |
|
| 数据集II[2M/1e-6形状,改变分类群(列)和基因(行)的数量] |
| 10 | 50 | 100 | 200 | 500 | 1000 |
|
| 50 | 0.3 ± 0.3 | 0.0 ± 0.1 | 0.3 ± 0.2 | 0.7 ± 0.3 | 6.0 ± 0.6 | 9.3±0.6 |
| 200 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.2 ± 0.09 | 3.9 ± 0.5 | 8.3 ± 0.5 |
| 1000 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.2 ± 0.08 | 1.7 ± 0.4 | |
| 数据集I[200分类群,不同树形(列)和基因数(行)]. |
|---|
| . | 10e-6(最近). | 10e-7(深). |
|---|
| 10米. | 200万. | 500公里. | 1000万. | 200万. | 500公里. |
|---|
| 50 | 0.2 ± 0.2 | 0.7 ± 0.3 | 37.9 ± 1.0 | 1.7±0.6 | 10.1 ± 0.9 | 38.7 ± 0.9 |
| 200 | 0.0 ± 0.1 | 0.2 ± 0.1 | 41.0 ± 1.1 | 0.7 ± 0.3 | 7.4 ± 0.7 | 41.4 ± 1.0 |
| 1000 | 0.0 ± 0.0 | 0.2 ± 0.1 | 39.2 ± 1.2 | 0.0 ± 0.0 | 3.5 ± 0.7 | 41.4 ± 1.1 |
|
| 数据集II[2M/1e-6形状,改变分类群(列)和基因(行)的数量] |
| 10 | 50 | 100 | 200 | 500 | 1000 |
|
| 50 | 0.3±0.3 | 0.0 ± 0.1 | 0.3 ± 0.2 | 0.7 ± 0.3 | 6.0 ± 0.6 | 9.3 ± 0.6 |
| 200 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.2 ± 0.09 | 3.9 ± 0.5 | 8.3 ± 0.5 |
| 1000 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.2±0.08 | 1.7 ± 0.4 | |
表1。与ASTRAL-I相比,ASTRAL-II获得的物种树误差减少
| 数据集I[200分类群,不同树形(列)和基因数(行)]. |
|---|
| . | 10e-6(最近). | 10e-7(深). |
|---|
| 1000万. | 200万. | 500公里. | 1000万. | 200万. | 500公里. |
|---|
| 50 | 0.2 ± 0.2 | 0.7 ± 0.3 | 37.9 ± 1.0 | 1.7 ± 0.6 | 10.1 ± 0.9 | 38.7 ± 0.9 |
| 200 | 0.0 ± 0.1 | 0.2 ± 0.1 | 41.0 ± 1.1 | 0.7 ± 0.3 | 7.4 ± 0.7 | 41.4±1.0 |
| 1000 | 0.0 ± 0.0 | 0.2 ± 0.1 | 39.2 ± 1.2 | 0.0 ± 0.0 | 3.5 ± 0.7 | 41.4 ± 1.1 |
|
| 数据集II[2M/1e-6形状,改变分类群(列)和基因(行)的数量] |
| 10 | 50 | 100 | 200 | 500 | 1000 |
|
| 50 | 0.3 ± 0.3 | 0.0 ± 0.1 | 0.3 ± 0.2 | 0.7 ± 0.3 | 6.0 ± 0.6 | 9.3 ± 0.6 |
| 200 | 0.0 ± 0.0 | 0.0±0.0 | 0.0 ± 0.0 | 0.2 ± 0.09 | 3.9 ± 0.5 | 8.3 ± 0.5 |
| 1000 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0±0.0 | 0.2 ± 0.08 | 1.7 ± 0.4 | |
| 数据集I[200分类群,不同树形(列)和基因数(行)]. |
|---|
| . | 10e-6(最近). | 10e-7(深). |
|---|
| 1000万. | 200万. | 500公里. | 1000万. | 200万. | 500公里. |
|---|
| 50 | 0.2 ± 0.2 | 0.7 ± 0.3 | 37.9 ± 1.0 | 1.7 ± 0.6 | 10.1 ± 0.9 | 38.7 ± 0.9 |
| 200 | 0.0±0.1 | 0.2 ± 0.1 | 41.0 ± 1.1 | 0.7 ± 0.3 | 7.4 ± 0.7 | 41.4 ± 1.0 |
| 1000 | 0.0 ± 0.0 | 0.2 ± 0.1 | 39.2 ± 1.2 | 0.0 ± 0.0 | 3.5 ± 0.7 | 41.4 ± 1.1 |
|
| 数据集II[2M/1e-6形状,改变分类群(列)和基因(行)的数量] |
| 10 | 50 | 100 | 200 | 500 | 1000 |
|
| 50 | 0.3 ± 0.3 | 0.0 ± 0.1 | 0.3±0.2 | 0.7 ± 0.3 | 6.0 ± 0.6 | 9.3 ± 0.6 |
| 200 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.2±0.09 | 3.9 ± 0.5 | 8.3 ± 0.5 |
| 1000 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.2 ± 0.08 | 1.7 ± 0.4 | |
ASTRAL-II的改进是由于增加了搜索空间。因此,我们询问用于向集合X添加双分区的启发式方法是否足够,或者通过进一步扩展X可以获得改进。为了回答这个问题,我们测试了将物种树中的所有双分区添加到集合X的影响,并比较了ASTRAL-II有无这些额外的双分区(参见补充图S2和补充数据). 我们发现ASTRAL-II在有和没有这些潜在的新双分区之间没有显著差异(P(P)=0.77,根据双向方差分析测试),表明ASTRAL-II的准确性不太可能通过扩大搜索空间进一步提高。
5.1.2运行时间
有200个分类群和较低水平的ILS,ASTRAL-I和ASTRAL-II的运行时间相似(补充图S2)但随着ILS的增加,ASTRAL-II速度更快(中位运行时间为3小时,而不是7.5小时)。请注意,ASTRAL-II搜索的树空间比ASTRAL-I大。由于分类单元数量较少,这两个版本的运行时间相近,但随着分类单元数量的增加,ASTRAL-II的运行时间增加得更慢(补充图S3). 对于500个分类群,ASTRAL-II的速度是ASTRAL-I的两倍(中位数为5小时对10小时),而ASTRAL-1在1000个分类群和1000个基因上没有完成。
5.2 RQ2:ASTRAL-II与其他聚合方法
此后,我们将ASTRAL-II称为ASTRAL。
在时间限制内完成ASTRAL在所有模型条件下完成,MP-EST仅在数据集上完成,除具有1000个基因和1000个分类群的条件外,所有模型条件最多完成100个分类群和NJst。
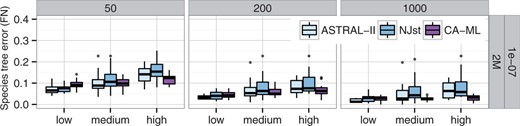
数据集IASTRAL在所有模型条件下都比NJst更准确,除了1e-07/500 K,这两种方法具有相同的准确度(图2). 总的来说,ASTRAL-II和NJst之间的差异具有统计学意义()根据双向方差分析检验,这些方法的相对性能受到物种形成率的显著影响(P(P)=0.026),但不取决于基因数量或树长。ASTRAL的速度比NJst快,在某些情况下是一个数量级,而在其他情况下是更小的幅度(补充图S4)。
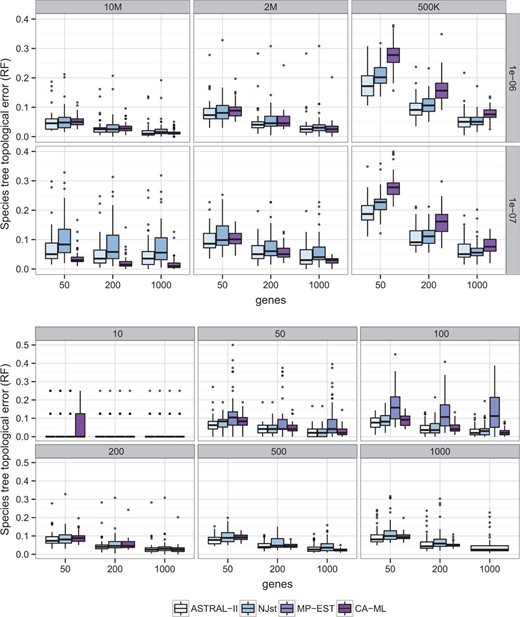
图2。
物种树拓扑精度的方法比较。(上图)200个分类群和不同的树形和基因数量。(底部)不同数量的分类群和基因,树形固定为2M/1e-6。ASTRAL-II至少与NJst和MP-EST一样准确
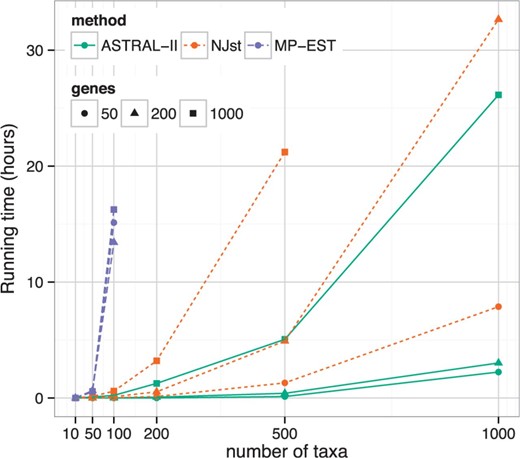
数据集II在10个分类单元数据集上,所有方法都具有较高的准确性(补充表S2). 在50个和100个分类单元的数据集上,MP-EST能够完成,但它是所有方法中最不准确的。ASTRAL在所有条件下都比NJst更准确,除了50个具有50个基因的分类群(补充表S2); 然而,当分类群数量为200个或更少时,差异通常较小,而当分类群更多时,差异更大。总的来说,ASTRAL和NJst之间的差异是显著的(P(P)=0.0007),并受到分类群数量的显著影响(P(P)=0.0004),但不包括基因数量。ASTRAL也比NJst快,特别是有更多的基因和更多的分类群(图3). 例如,在500个分类群和1000个基因上,ASTRAL通常在2-10小时内完成,而NJst需要12-30小时(补充图S5). MP-EST是迄今为止最慢的方法,但其运行时间不受基因数量的影响。
图3。
与不同数量的分类群和基因的运行时间比较(数据集II)显示了NJst和ASTRAL-II的平均运行时间。请注意,ASTRAL-II在大型数据集上要快得多
5.3 RQ3:ASTRAL-II与CA-ML
5.3.1数据集I
有趣的是,树长对CA-ML和ASTRAL的相对准确性有显著影响(),物种形成率(P(P)=0.00004)和基因数量(). 在ILS水平较低(10M和2M)和最近物种形成的情况下,CA-ML和ASTRAL具有相近的准确性,但CA-ML往往随着基因的增加而更好,ASTRAL随着基因的减少而更好(补充表S1,图2). 随着物种形成的深入和ILS的降低,CA-ML比ASTRAL更准确,但增加基因数量可以缩小差距。在高ILS水平下,ASTRAL对所有数量的基因以及最近和深层物种形成来说都比CA-ML准确得多。
5.3.2数据集II
总的来说,ASTRAL和CA-ML之间的差异不显著(P(P)=0.2),但相对准确性似乎受到基因数量的影响(P(P)= 0.06). 无论分类群的数量如何,这都不会影响相对准确性(P(P)=0.2),1000个基因的CA-ML略为准确,基因较少的ASTRAL略为精确(补充表S2,图2)。
5.3.3运行时间
我们在不同的平台上运行CA-ML和ASTRAL-II,因此无法进行直接的运行时间比较。尽管如此,我们还是提供了运行时间数字,以便大致了解情况。在含有1000个基因的200轴模型条件下,使用FastTree进行CA-ML大约需要2小时,而ASTRAL-II估计物种树和估计基因树也需要大约1.5小时,因此,ASTRAL-III和CA-ML的运行时间在该数据集上相对接近。
5.4 RQ4:基因树错误的影响
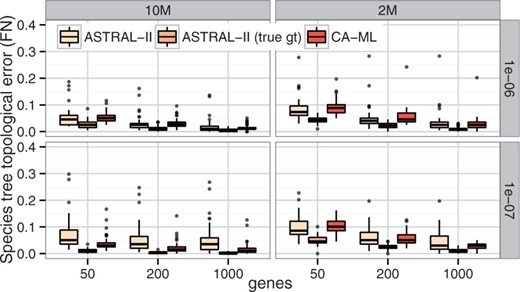
在RQ3中,我们观察到,在某些条件下,CA-ML比ASTRAL更准确,我们将这种模式归因于模拟中存在的高水平基因树错误。当使用真(模拟)基因树而不是估计的基因树时,无论模型条件如何,ASTRAL的准确性都很高(参见图4和补充图S6),ASTRAL总是比CA-ML更准确。因此,在ILS较低水平下,CA-ML比ASTRAL更准确的事实与提供给ASTRAL的输入中的估计误差有关。
图4。
使用估计和真实基因树以及数据集I上的CA-ML进行ASTRAL-II运行的比较
在我们的ASTRAL和NJst分析中,基因树误差与物种树误差呈正相关(补充图S7)相关系数与ASTRAL和NJst相似。CA-ML的误差也与基因树误差相关(显然,这种关系是间接的;例如,短比对同时影响CA-ML和基因树误差),但相关性弱于基于联合方法的相关性(补充图S8). 有趣的是,基因树估计误差和物种树误差之间的相关性通常在基因较少的情况下较高。
为了进一步研究基因树误差的影响,我们将每个模型条件的重复次数分为三类:平均基因树估计误差低于0.25为低,介于0.25和0.4之间为中等,高于0.4为高。我们在这些类别中绘制了物种树的精确度(参见图5对于一种模型条件,另请参见补充图S9和补充数据对于其他模型条件)。ASTRAL和NJst的相对性能在不同类别的基因树错误中通常保持不变,但基因树错误的增加往往会增加ASTRAL与NJst之间的差异程度。此外,MP-EST似乎比NJst或ASTRAL对基因树错误更敏感(补充图S10)。
图5。
比较种树精度与200个分类群,将基因树估计误差分为三类。方框显示基因数量
ASTRAL和CA-ML的相对性能取决于基因树错误。对于那些CA-ML通常比ASTRAL更准确的模型条件(例如2 M/1e-07),ASTRAL在基因树估计误差较低的重复上往往优于CA-ML(图5). 与此一致,我们注意到ASTRAL受基因树错误的影响大于CA-ML(补充图S9)。
5.5 RQ5:坍塌低支撑分支
ASTRAL-II可以处理多段输入。虽然我们还没有进行引导以获得可靠的支持措施,但我们确实从FastTree-II获得了本地SH-like分支支持。我们折叠了低支持度分支(10%、33%和50%),并对产生的未解析基因树运行ASTRAL。我们测量了收缩低支撑分支对射频率的影响:中位数增量射频(崩溃前的误差减去崩溃后的误差)通常为零(补充图S11),从不高于零,但在少数情况下低于零(表示在这些情况下准确性有所提高)。然而,这些差异在统计学上并不显著(P(P)= 0.36). 由于该分析是使用类似SH的分支支持值而不是bootstrap支持值(或其他估计支持值的方法)进行的,因此需要进一步研究。
6生物学结果
被子植物的进化和毛足Amborella trichopoda Baill。是陆地植物进化中具有挑战性的问题之一。最近的一些分子研究中恢复了一个假设(例如。德鲁等。2014;邱等。2000;威基特等。2014;张等。2012)是那个吗答:。毛足类。是其他被子植物的姊妹植物,其次是睡莲(即睡莲目)。特别是,最近根据整个转录组对104种植物进行了分析,利用数据集的各种扰动,通过串联和ASTRAL-I恢复了这种关系(威克特等。, 2014). 一个相互矛盾的假设是无油樟是睡莲的姊妹,而这整个群体是其他被子植物的姊妹(德鲁等。, 2014;戈列梅金等。, 2013)。Xi(希)等。(2014)使用从42个被子植物和4个外群中采集的310个基因来研究这个问题。他们观察到CA-ML产生了第一个假设,MP-EST产生了第二个假设,他们认为这些差异是由于CA-ML不建模ILS,而MP-EST建模ILS。
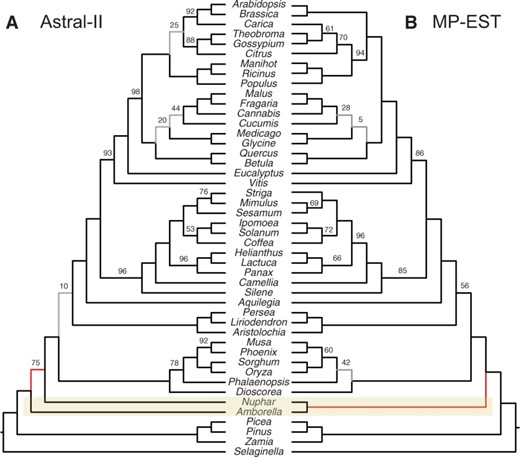
我们从Xi(希)等。(2014)以及在GTR+Γ模型下使用RAxML估计基因树,具有200次重复的自举和10轮ML(使用RAxML是因为在这个相对较小的数据集上运行时间不是问题)。我们运行了MP-EST和ASTRAL,得到了两棵不同的树(图6). 正在复制Xi(希)等。(2014)结果,MP-EST恢复了无油樟和Nymphaeales获得100%支持。然而,与CA-ML一样,ASTRAL也能恢复无油樟作为其他被子植物的姊妹,得到75%的支持。尽管无油樟我们的分析表明,CA-ML和MP-EST结果之间的差异不能简单地归因于CA-ML不考虑ILS这一事实。
图6。
基于被子植物数据集计算的物种树的比较 Xi(希)等。(2014).MP-EST和ASTRAL-II在无油樟; 连接树符合ASTRAL-II
7讨论和结论
我们的广泛模拟结果表明,与我们研究的其他方法不同,ASTRAL-II可以在合理的运行时间内分析多达1000个分类群和1000个基因的数据集。然而,未来的研究需要将ASTRAL-II与分而治之的方法进行比较(例如。拜齐德等。, 2014;齐默尔曼等。, 2014)这使得较慢的基于聚合的方法能够扩展到大型数据集。ASTRAL-II比其他基于聚合的方法更准确,也比CA-ML更准确,除非ILS水平低,基因树误差高。虽然我们研究的被子植物生物数据集相对较小(46种),但我们的模拟表明,可以使用ASTRAL-II准确分析即将出现的具有大量物种的多基因数据集。
在被子植物数据集上,ASTRAL恢复了CA-ML和大量近期研究支持的关系,而MP-EST恢复了一个替代拓扑,也得到了一些先前分析的支持。这两种方法之间的差异可能有几个原因,包括生根基因树(MP-EST要求,ASTRAL不要求)的可能性卷柏属对于某些基因来说可能存在问题,或者对于这两种方法,基因树估计误差的影响是不同的。我们还注意到,ASTRAL是一种不估计分支长度的非参数方法,与参数方法(如MP-EST)相比,非参数方法可能对基因树估计误差不太敏感。
ASTRAL比CA-ML更准确,除非基因树估计误差较大且ILS水平足够低。这些结果表明,不应拒绝CA-ML,即使在多物种合并模型下它在统计上不一致。相反,标准汇总方法一致性的证明假设基因树估计无误(Roch和Warnow,2015年)这种假设限制了一致性结果在实践中的相关性。如文献(例如。米拉布等。2014年b,c(c);帕特尔等。2013); 然而,使用无重组区域的要求使这种追求复杂化,因为无重组“c基因”可能很短,特别是随着分类群数量的增加(盖茨·斯普林格(Gatesy and Springer),2014年). 未来的研究需要研究使用更短的基因序列比对的影响,反之,基因内重组事件的存在,用作基于合并的物种树估计方法的输入。
确认
我们感谢匿名评论员提出的有益建议。
基金
这项工作得到了国家科学基金会的支持[0733029、1461364和1062335(给T.W.)];霍华德·休斯医学院(HHMI)研究生奖学金(发给S.M.)。
利益冲突:未声明。
工具书类
等(
2014
)磁盘覆盖方法改进了系统发育分析
.BMC基因组学
,15
(补充6
),第7部分
.
(
2009
)基因树不一致、系统发育推断和多物种融合
.经济趋势。进化。
,24
,332
——340
.
等(
2014
)对被子植物根部的另一次观察揭示了一个熟悉的故事
.系统。生物。
,63
,368
——382
.
(
2009
)新的一般分子系统学理论正在兴起吗?
进化
,63
,1
——19
.
(
2009
)不可磨灭:生物序列进化的灵活模拟器
.分子生物学。进化。
,26
,1879
——1888
.
(
2014
)深层时间尺度的系统发育分析:不可靠的基因树,绕过隐藏支持,以及聚合/连接难题
.分子系统发育。进化。
,80
,231
——266
.
等(
2013
)开花植物的进化根
.系统。生物。
,62
,50
——61
.
(
2010
)基于多点数据的物种树贝叶斯推断
.分子生物学。进化。
,27
,570
——580
.
(
2007
)合并条件下连锁数据中系统发育估计的不一致性
.系统。生物。
,56
,17
——24
.
等(
2011
)RAxML和FastTree:两种大规模最大似然系统发育估计方法的比较
.公共科学图书馆一号
,6
,e27731号
.
等(
2010
)合并模型下估计种树的最大伪似然方法
.BMC演变。生物。
,10
,302
.
(
2011
)从未生根的基因树中估算物种树
.系统。生物。
,60
,661
——667
.
(
1997
)物种树中的基因树
.系统。生物。
,46
,523
——536
.
等(
2014年a
)ASTRAL:基于基因组尺度合并的物种树估计
.生物信息学
,30
,i541型
——i548型
.
等(
2014年b
)不完全谱系排序下多位点物种树估计的总结方法评价
.系统。生物。
,pii公司
,.
等(
2014年c
)统计装箱可以准确地对鸟类树进行基于合并的估计
.科学类
,346
,1250463
.
等(
2013
)生命树中灌木的系统发育估计错误
.《谱系学杂志》。进化。生物。
,1
,110
.
等(
2010
)FastTree-2用于大型比对的近似最大似然树
.公共科学图书馆一号
,5
,电子9490
.
等(
2000
)基础被子植物的系统发育:三个基因组中五个基因的分析
.国际植物科学杂志。
,161
,第3章
——S27系列
.
(
2003
)基于多基因座DNA序列的物种分化时间和祖先种群规模的Bayes估计
.遗传学
,164
,1645
——1656
.
(
1981
)系统发育树的比较
.数学。Biosci公司。
,53
,131
——147
.
(
2014
)在对齐序列数据集的串联上基于似然的树重建在统计上可能不一致
.西奥。大众。生物。
,100
,56
——62
.
(
2015
)基于合并的物种树方法对基因树估计误差(或缺乏)的鲁棒性
.系统。生物。
,pii公司
,syv016型
.
(
2014
)RAxML第8版:大系统发育分析和后分析工具
.生物信息学
,30
,1312
——1313
.
等(
2014
)陆地植物起源和早期多样性的植物学分析
.程序。国家。阿卡德。科学。美国
,111
,E4859型
——E4868型
.
等(
2014
)结合与串联方法以及无油樟作为睡莲的姐姐
.系统。生物。
,63
,919
——932
.
等(
2012
)高度保守的低拷贝核基因作为被子植物系统发育分析的有效标记
.新植物醇。
,195
,923
——937
.
等(
2014
)BBCA:使用随机装箱提高*BEAST的可扩展性
.BMC基因组学
,15
(补充6
),第11节
.
©作者2015。牛津大学出版社出版。
这是一篇根据知识共享署名非商业许可条款分发的开放获取文章(http://creativecommons.org/licenses/by-nc/4.0/)它允许在任何媒体上进行非商业性重复使用、分发和复制,前提是正确引用了原始作品。如需商业再使用,请联系journals.permissions@oup.com








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}