技术报告
研究疾病的基因组方法的一个优势是用无偏见但通用的假设取代了知情但有偏见的假设,例如在全基因组关联研究(GWAS)中“平等对待”所有遗传变异。然而,对于大效应的罕见变体和弱效应的常见变体,使用先验知识对疾病基因发现至关重要1–4例如,外显子组测序是一种有效的发现策略,因为它专注于蛋白质改变变异,丰富了因果效应5.
虽然许多现有注释有助于确定因果变量的优先级,以提高发现能力(例如PolyPhen6,筛选7和GERP8),当前的方法往往受到四个主要限制中的一个或多个限制。首先,注释在输入和输出方面差异很大。例如,保护指标8–10是全基因组定义的,但不使用功能信息,也不是等位基因特异性的,而基于蛋白质的指标6,7仅适用于编码变异,通常仅适用于错义变异,因此排除了99%以上的人类遗传变异。其次,每个注释都有自己的度量标准,而这些度量标准很少具有可比性,因此很难评估不同变体类别或注释的相对重要性。第三,对已知致病性突变进行训练的注释会受到主要的确定偏差的影响,并且可能不会泛化。第四,获取,更不用说客观地评估或组合现有的部分相关和部分重叠注释的全貌,是一个重大的实际挑战;这一挑战只会随着像ENCODE这样的大型项目而扩大11不断增加可用的相关数据量。这些限制的最终结果是,许多潜在的相关注释被忽略,而使用的子集被应用并组合在特别的以及破坏其效用的主观方式。
在这里,我们描述了一个通用框架,即组合注释依赖性缺失(CADD),用于整合不同的基因组注释并对任何可能的人类单核苷酸变异(SNV)或小插入/缺失(indel)事件进行评分。CADD的基础是对比人类相对于模拟变异体的固定或近固定衍生等位基因注释。有害变种——即降低生物体适应性的变种——被固定但非模拟变种的自然选择耗尽。因此,CADD测量有害性,这一特性与分子功能和致病性密切相关12重要的是,与致病性或分子功能性相比,有害性指标具有主要优势。虽然后者的范围仅限于一小组遗传或实验上特征明确的突变,并且受到主要的确定偏差的影响,但有害性可以在整个基因组集合中进行系统测量(参见参考文献8,9,10和以下)。此外,对遗传变异的选择性约束与它们的表型相关效应的总和有关,而不是与任何单个分子或表型结果有关。因此,原则上,毒性测量可以提供对变异影响的全基因组、数据丰富、功能通用和组织相关的估计。
我们确定了人类基因组和推断出的人类黑猩猩祖先基因组之间的差异13其中人类携带的衍生等位基因频率至少为95%(1490万SNV和170万indels)。几乎所有这些事件在人类血统中都是完全固定的,在1000基因组项目中,只有不到5%的事件表现为几乎固定的多态性14变异目录(衍生等位基因频率(DAF)≥95%)。要模拟等效数量的从头开始的突变,我们使用了序列进化的经验模型,其中CpG二核苷酸特异性比率和突变率在1兆碱基(Mb)尺度下局部估计(补充说明). 通过六向灵长类基因组比对估计了indels的突变率参数和大小分布15.
为了生成注释,我们使用了Ensembl Variant Effect Predictor16(VEP),来自ENCODE项目的数据11和来自UCSC基因组浏览器轨迹的信息17(补充表1). 注释涵盖了一系列数据类型,包括GERP等保护指标8,相位cons9和phyloP10; 监管信息11DNA酶超敏反应的类基因组区域18和转录因子结合19; 转录信息,如到外显子-外显子边界的距离或在常用研究细胞系中的表达水平11; 和蛋白质水平得分,如格兰瑟姆20、SIFT7,和聚Phen6得到的逐注释方差矩阵包含2940万个变体(半固定或接近固定的人类衍生等位基因(“观察到的”),半模拟从头开始的突变(“模拟”)和63个不同的注释,其中一些是总结许多底层注释的复合注释(补充说明,补充表1-2).
我们首先通过构建一系列单变量模型来评估我们的通用方法的有效性,这些模型将63个注释中的每一个作为单独的预测因子来对比观察到的和模拟的变量(补充说明). 几乎所有模型都非常重要(补充表3-5)与预期一致。例如,我们发现无义变异体耗竭了近20倍,错义变异物耗竭了2倍,基因间或上游/下游变异体没有耗竭(补充表6). 发生在cDNA起始点附近的无义和错义突变比发生在末端附近的突变耗竭得多(补充表7),并且20内,特别是2内的变异,剪接连接的核苷酸也被耗尽(补充图1). 表现最好的单个注释是蛋白质水平的度量,如PolyPhen6和SIFT7,但这些仅评估了错义变体(训练数据中0.63%的所有变体是错义的;其中88%定义了PolyPhen值,90%定义了SIFT值)。保护指标是最强的个体全基因组注释(补充表3).
我们还检查了注释之间的相关性(补充图2)以及在注释之间添加交互项的价值(补充图3). 许多注释是相关的,许多交互作用在统计上是显著的,但只有少数交互对有意义地改进了简单的加性模型。总的来说,这些分析表明,相对于63个注释,观察到的变体和模拟的变体之间存在着重大的生物学差异,线性模型捕获了大部分这类信息。
接下来我们训练了一个支持向量机21(SVM),基于从63个注释派生的特征的线性核,并辅以有限数量的交互项(补充说明,补充表1-2,补充图4). 根据观察到的变异和模拟变异的不同样本独立训练的10个模型高度相关(所有配对Spearman秩相关>0.99;补充图5). 应用这些模型的平均值对人类参考基因组(GRCh37)中86亿个可能的SNV进行评分。为了简化某些上下文中的解释,我们还定义了phred-like22分数(“量表C分数”)基于每个变量相对于86亿SNV的C分数排名,范围从1到99(补充说明). 例如,最高10%(10−1)在所有得分中,也就是说,在我们的模型下,最不可能观察到的人类等位基因被分配为10或更大的值(“≥C10”),而变异最高的1%(10−2), 0.1% (10−3)等被分配得分≥C20、≥C30等。
我们首先计算了具有特定功能后果的给定量表C评分的所有可能替代的比例(图1;补充表8). 尽管仅根据观察到的变异和模拟变异之间的差异进行训练,而不是根据可能引入确定偏差的已知致病变异集进行训练,但潜在无义变异的C核最高(中位数37),其次是错义和规范剪接位点变异(中位数15)基因间变异位于列表底部(中位数2)。同时,76%≥C20的潜在SNV是非编码的(即而74%的潜在错义SNV和18%的潜在无义SNV低于C20。此外,在每个功能类中,都有一些生物学上相关的区别,并且可能具有预测价值。例如,在疾病研究中,嗅觉受体中的潜在无义变体通常被视为一个同质组,其得分低于其他基因,而之前发现的基因中的潜在无义变体是“必需的”23得分更高(图1下部面板,补充图6). 因此,C分数可以捕获功能类别之间和内部的大量信息。值得注意的是,由于缺失(例如,仅针对错义措施)或缺乏功能意识(例如,保护措施无法区分给定位置的无义和错义等位基因),这些相同的区别在其他措施中不存在或减弱。
图1。
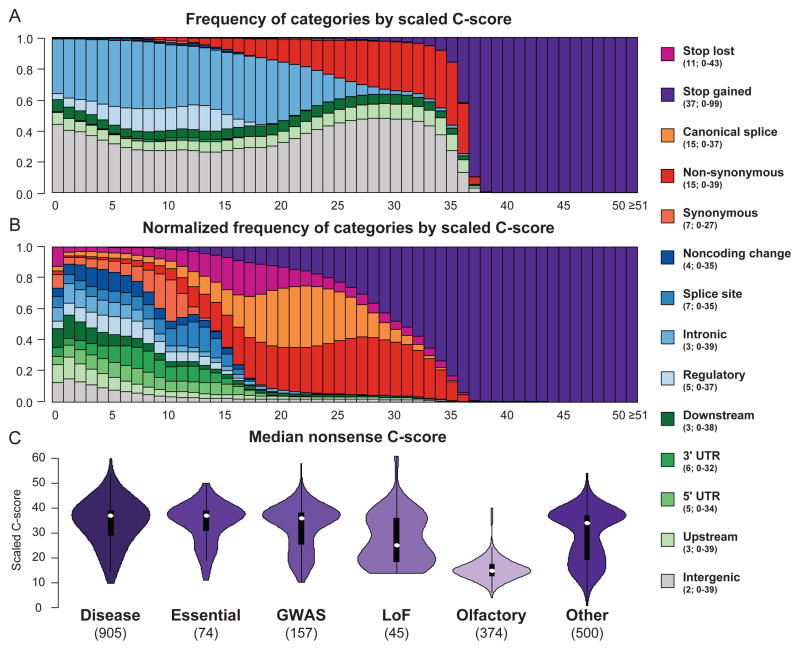
量表C评分与分类变量结果的关系。上面的图显示了每个标度C分值箱中具有特定结果的替换的比例,而中间的面板显示了通过该类别中观察到的变体总数进行首次标准化后具有特定结果替换的比例。图例显示了每个类别按比例计算的C分值的中位数和范围。结果从集合变量效应预测器中获得16(补充说明)例如,“非编码变更”是指带注释的非编码转录本中的变更。每个C评分箱中功能分配的详细计数见补充表8下面板显示了包含至少5种已知致病性突变的基因的潜在无义(停止着色)变体的中位C分数的小提琴图48(“疾病”);被预测为“基本”23; 与复杂性状相关的港口变异41(“GWAS”);在1000个基因组中至少有2个功能丧失突变49(“LoF”);编码嗅觉受体蛋白;或随机选择500个基因(“其他”;参见补充说明).
接下来,我们将量表C评分与遗传多样性水平进行了比较,发现C评分与1000基因组项目中确定的变异体的DAF呈负相关14或Exome测序项目24(电子稳定程序)(图2a;补充图7–9),1000基因组项目目录中人类遗传变异的减少(图2b),以及黑猩猩衍生变体的缺失(图2c). 重要的是,这些验证数据集与训练数据的“观察”子集重叠最小,训练数据仅包含固定或几乎固定(>95%DAF)的人类衍生等位基因。此外,尽管我们不能完全消除这些因素的混淆,但C分数和站立变异的DAF之间的负相关对于控制背景选择、局部GC含量、局部CpG密度和基于位置的保守性的变化是稳健的(补充图9).
图2。
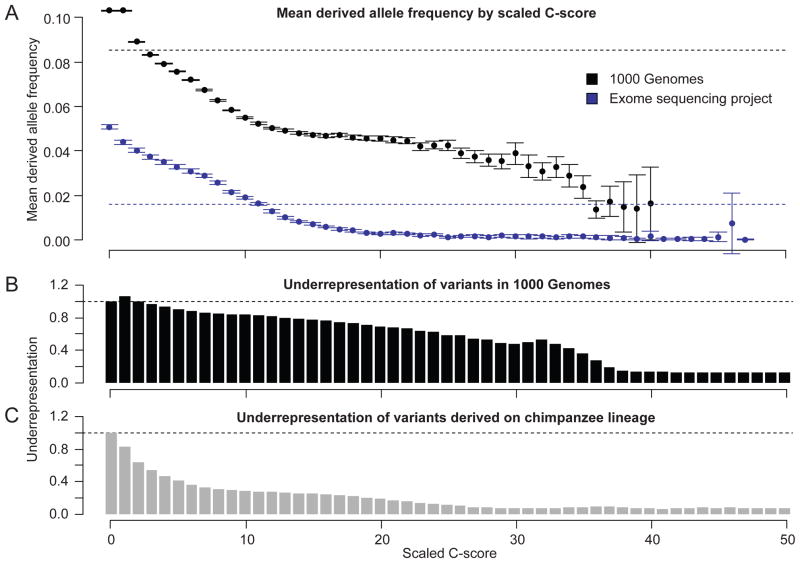
量表C分值与:1000基因组项目中确定的变异的平均衍生等位基因频率(DAF)之间的关系14或ESP24(上面板);1000个基因组多态位点的低表达(中间面板);和黑猩猩血统衍生变种(下表)。上图中的虚线表示平均DAF,置信区间表示每个箱子中平均(SEM)DAF的1.96倍标准误差。低分辨率定义为1000个基因组(中间面板)或黑猩猩衍生(下部面板)的比例特定标度C评分箱中的变体除以观察人类参考集合所有可能突变的标度C得分的频率(10C分数/−10). 与1000个基因组变异体相比,黑猩猩衍生变异体的低分辨率预计更强,因为前者大多是固定或高频变异体(并经历了多代纯化选择),而后者大多是低频变异体。除0分外,两个面板的C分箱消耗值与预期值存在显著差异(二项式比例测试,所有p值<10−11).
接下来,我们试图评估CADD在五种不同情况下优先考虑功能和疾病相关变异的效用。
首先,针对MLL2级是歌舞伎综合征中突变的基因,C评分可以区分多种疾病相关等位基因25与罕见的、可能是良性的ESP变异相比24(威尔科森秩和检验p=9.9×10−94; n=210/679)。其他指标在准确性或全面性方面明显较差(补充图10).
其次,对于乙型肝炎病毒、β地中海贫血基因突变、疾病相关等位基因C评分26–一组具有调控/上游(n=54)、剪接(n=37)、错义(n=22)、无义(n=18)和其他影响的indels(n=93)和SNVs(n=119)与三种表型严重程度相关,且比其他指标更为显著(Kruskal-Wallis秩和检验p=2.4×10−7; n=48/65/99,补充图11).
第三,NIH ClinVar数据库管理的致病性变体27与可能的良性等位基因(ESP24DAF≥5%)与相同的分类结果相匹配(Wilcoxon秩和检验p<10−300,n=8174/8174,图3;补充图12-16). 我们注意到ClinVar和PolyPhen基础训练数据之间存在大量重叠。当这些位点被排除在测试数据集之外,或者当PolyPhen被排除在CADD的训练特征之外时,C核继续优于所有或几乎所有仅错义的指标和保护措施(补充图12).
图3。
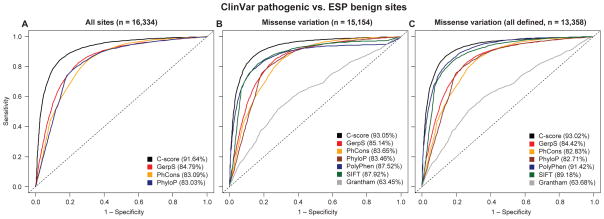
用于区分NIH ClinVar数据库定义的精选致病性突变的受体操作特征(ROC)27与明显良性的ESP等位基因匹配(DAF≥5%)24具有相同的分类结果。左侧面板显示了定义了GerpS、PhCons和PhyloP评分的全基因组变异(n=16334),而中间面板将分析限制在错义变化(n=15154),缺失值被估算到每个评分的上限,右侧面板显示错义变化,其中PolyPhen,SIFT和Grantham分数均已确定(n=13358)。右侧面板的版本不包括PolyPhen训练数据和ClinVar数据库之间的重叠,或使用未使用PolyPhon训练的CADD模型作为特征,如所示补充图12曲线下面积(AUC)值在图图例中提供了所使用的每个分数。
第四,C评分与向国际癌症研究机构报告的p53体细胞癌突变观察数密切相关(斯皮尔曼等级相关0.38,p=6×10−73,n=2068,补充说明).
第五,我们检测了两种增强剂28和一个启动子29在此之前,我们进行了饱和突变。C-分数与实验测量的个别变异的绝对表达倍数变化显著相关,总体上比序列保守性更为相关(综合数据的Spearman秩相关=0.31,p=1.9×10−65,n=2847;补充图17).
总的来说,这些分析表明,CADD在各种实验和疾病环境中定量预测了蛋白质改变和调节的有害性、致病性和分子功能。在每一种情况下,CADD的预测效用都比序列守恒的测量要好得多,序列守恒是唯一一种综合类型的变体得分,而且在大多数情况下,当限制在适当的变体子集时,它往往比函数特定的度量好得多。
接下来,我们考虑了CADD如何在外显子组或全基因组研究中评估候选变异。
首先,我们分析了从头开始的自闭症谱系障碍儿童的外显子变异体(SNV和indels)30–34(ASD)和智力残疾35,36(ID)以及未受影响的同胞或对照,包括88个无义词、1015个错义词、359个同义词、32个规范剪接位点和150个其他变体,包括indels。单独考虑每种疾病,受影响儿童的变异比未受影响的兄弟姐妹/对照组的变异有害得多(补充表9)或组合(ASD+ID Wilcoxon秩和检验p=2.0×10−4,n=1130/514)。此外,从头开始的ID先证者的变异比ASD先证者更有害(p=4.7×10−5,n=170/960),表明ID中存在更有害的全局突变负担,这与ID中相对于ASD的拷贝数变体的大小和数量增加的观察结果一致37.
第二,众所周知,像PolyPhen和conservation这样的注释在基于序列的疾病基因识别中很有价值,因为它们能够对致病性变体进行高度排序1,2,38因此,我们检查了代表不同人群的11个个体基因组中的C得分分布39,40,并发现CADD在个人基因组的全谱变异中对已知的致病性变体(ClinVar致病性)进行了高度排名(图4;补充图16和补充表10-11). 此外,CADD在这项任务中更加定量和全面(例如,约27%的致病性ClinVar SNV未被PolyPhen评分,因为缺失值或其对错义变异的限制)。鉴于CADD在个人基因组内的全谱变异中对已知致病性变体进行排序方面,相对于现有的最佳蛋白质和保守性指标具有相当大的优势,它很可能会提高基于序列的疾病研究的能力,超越当前的标准方法。
图4。
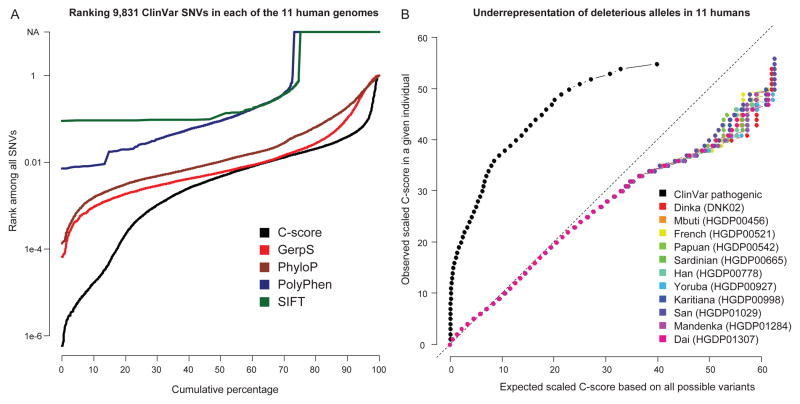
对来自不同人群的11名人类个体进行全基因组测序,确定致病性ClinVar变异体在变异体中的排名。左侧面板:11个个人基因组中每一个的9831个致病性ClinVar变异体“突然出现”时的等级累积分布。例如,约30%的ClinVar变体的C-核心在个人基因组中的所有变体中排名前0.1%,大多数变体排名前1%。约25%的致病性ClinVar SNV未被PolyPhen/SIFT评分,因为缺失值或其对错义变异的限制;还请注意,PolyPhen/SIFT的等级仅在错义变体中计算,因此从总变体中派生出来的数量要少得多(参见补充图16). 右侧面板:11名患者和致病性ClinVar SNV的SNV C评分QQ图。对于在个体中观察到的给定标度C分数,计算出该个体C分数至少如此大的变体的分数(y轴)。与所有可能变量的分布分位数相对应的C分数显示在x轴上。与所有可能的变体组相比,高C分数代表性不足。相反,来自ClinVar的已知病媒变异体相对于所有可能变异体的集合具有较大的C评分。可以利用这一事实优先考虑从单个基因组的全基因组测序中确定的因果变异(左面板和补充表10–11).
最后,我们分析了复杂性状GWAS鉴定的单核苷酸多态性(SNP)的CADD评分,并将其与与等位基因频率和基因分型阵列可用性匹配的邻近对照SNP进行了对比(图5,补充说明). 我们发现,领先的GWAS SNP的C得分显著高于对照SNP(单侧Wilcoxon秩和检验,p值=1.3×10−12,n=5498/5498);与领先SNP(“标签”)连锁不平衡的邻近SNP平均得分低于领先SNP,但也显著高于其匹配对照(p值=5.1×10−107). 在控制了基因体效应、基因表达水平、保守性和调节元件重叠等特性后,C-核心差异仍然显著;相关SNP和对照SNP之间的每一个都有显著差异,但没有一个能完全解释C评分差异(补充说明). trait-associated SNPs的C分值进一步与潜在关联研究的规模以及关联本身的统计意义相关(图5;补充图16;补充说明)这可能是由于更大的研究和更强的关联统计数据增加了对因果变量的丰富能力。虽然在大多数情况下并非因果关系,但我们的分析表明,GWAS识别的SNP,尤其是来自大型研究的强关联主SNP,在因果变异方面得到了丰富,这与之前观察到的GWAS对单个注释的丰富一致11,41–44.
图5。
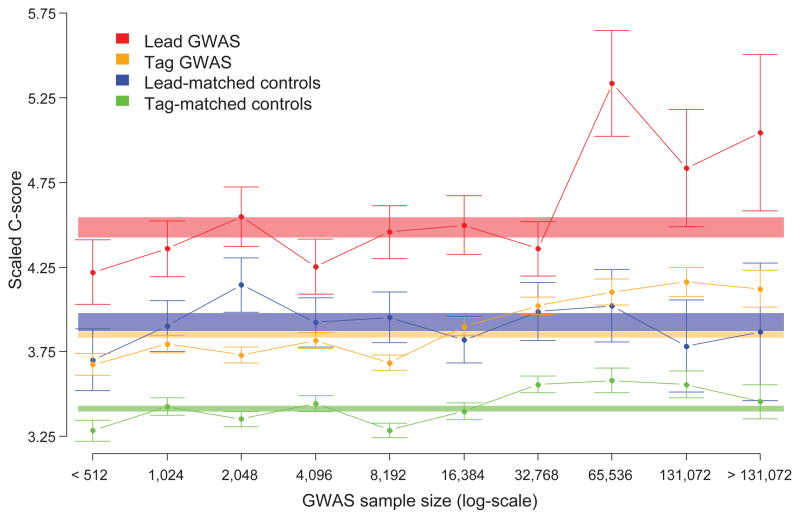
GWAS SNP的C分数高于附近的对照SNP,并且取决于研究样本的大小。根据识别SNP的关联研究的样本大小(x轴),绘制每类SNP的平均标度C得分(y轴),如颜色所示。样本量箱为原木2-规模化且相互排斥;例如,标记为“1024”的箱子代表512到1024个样本研究中的所有SNP。误差条是平均值的±1标准误差(SEM)。阴影矩形代表整体,即在所有样本大小中,按比例计算的C得分意味着每个类别的±1 SEM,如颜色所示。
通过CADD,我们描述了一个通用的、可扩展的框架,用于将遗传变异的各种注释中包含的信息集成到一个分数。我们证明,在各种情况下,这种方法在优先考虑功能性和致病性变体方面比其他广泛使用的注释更好,在某些情况下是适度的,但在许多情况下是显著的。此外,除了在任何一种情况下的效用外,CADD还有实际和概念上的优势,这应该被证明对人类疾病的遗传研究具有重要价值。首先,许多单个注释的信息内容被客观地合并为一个值,这比特别的组合注释并可能提高性能的方法,与特定于错义注释的“共识”方法的优点一致45其次,CADD可以很容易地将扩展合并到现有注释和全新注释中。鉴于像ENCODE这样的项目正在不断快速地扩展可用的注释,无限期地、轻松地集成新信息的能力至关重要11第三,CADD将保守性指标的一般性与子相关功能指标(如PolyPhen)的特殊性结合起来,利用两者的优点,同时削弱各自的缺点。
CADD也有一些限制,可能会限制其在某些分析中的实用性或代表需要改进的领域。首先,C评分衡量变异的减少,这与有害性相关,但也受局部突变率、背景选择、有偏见的基因转换和其他现象的影响,可能限制准确性。其次,C分数反映了具有给定注释模式的变体的比例,这些变体对选择可见,但可能无法捕捉到选择强度的差异;其他方法,如多态性-差异性比较,可能更准确地估计选择性系数46第三,人们强烈需要更多的“金标准”数据,尤其是基因组的非编码区域,目前的缺乏限制了更好注释的发展以及我们验证预测的能力。第四,目前不可能精确校准CADD估计的有害性与变体致病可能性之间的关系。因此,C评分最好根据“有害可能性”而不是“致病可能性”来解释,例如黑猩猩衍生等位基因给定C评分的可量化损耗程度(图2c,补充表11). 特别是在发现因果变异时,CADD应被视为一条有助于提供致病性证据的信息,并作为遗传信息的补充而非替代进行评估。
CADD的“一站式”性质可能对未来的测序研究具有很大的实用价值和概念价值。它将最小化必须由实验室或项目生成、跟踪和评估的注释的范围和多样性,并减少对特别的过滤器、分数和参数的组合,就像现在常规做的那样。例如,外显子组研究中一种常用的方法是在遗传分析之前,将错义(有或没有“损伤”注释或给定的保守性水平)、无义和剪接分离变体合并为一个内部未分类的“蛋白替代”变体列表5使用CADD,可以完全避免任意过滤器/阈值,包括在一个有意义的排序列表上的编码和非编码变体。例如,最近一项关于隐性非综合征性胰腺发育不全的研究确定了5种因果非编码变异体,它们破坏了胰腺癌远端增强子的功能PTF1A型47这些非编码疾病变异的C评分(标度评分在23.2到24.5之间)将其排在所有可能的人类SNV的99.5%以上,典型外显子中错义SNV的97%以上,以及ClinVar中孟德尔致病性SNV的56%以上27.
在研究和临床中,我们定义遗传变异目录的能力超过了系统评估其潜在影响的能力。随着测序速度的加快,基因组取代外显子,以及功能类别和注释阵列的扩大,这一挑战将加深。为了应对这一挑战,一个能够利用许多基因组注释的统一、定量和可扩展的框架至关重要。我们预计这里描述的模型以及随之而来的所有可能的GRCh37/hg19 SNV的免费预计算分数(http://cadd.gs.washington.edu/)将立即发挥广泛的作用,并随着时间的推移而不断改进,从而能够更好地解释临床环境中具有不确定意义的变体,并提高孟德尔病和复杂疾病遗传研究的发现能力。

