摘要
单核苷酸多态性(SNP)研究和随机诱变项目确定了蛋白质编码区的氨基酸取代。每一种替代都有可能影响蛋白质功能。SIFT公司(S公司运动我容错剂F类只读存储器T型olerant)是一个预测氨基酸替代是否影响蛋白质功能的程序,以便用户可以优先考虑替代以进行进一步研究。我们已经证明,SIFT可以在诱变研究和人类多态性研究中区分功能中性和有害氨基酸的变化。SIFT的网址为http://blocks.fhcrc.org/sift/sift.html.
收稿日期:2003年1月4日;修订并接受2003年2月28日
简介
单核苷酸多态性(SNP)被用作连锁和关联研究中的标记,以检测人类基因组中哪些区域可能与疾病有关。编码区和调控区的单核苷酸多态性可能与疾病本身有关。引起蛋白质产物中氨基酸变化的非同义SNP是人们最感兴趣的,因为氨基酸替换目前约占导致人类遗传病的已知基因损伤的一半(1). SIFT公司(S公司运动我容错剂F类只读存储器T型olerant)使用序列同源性预测氨基酸替换是否会影响蛋白质功能,从而可能改变表型(2,三).
SIFT已应用于人类变异数据库,并能够区分与疾病相关的突变和中性多态性(三). 假设致病氨基酸替换会损害蛋白质功能,我们将SIFT应用于与疾病相关或涉及疾病的错义替换数据库(4). SIFT预测69%具有破坏性。当SIFT应用于dbSNP中的非同义SNP时(5)据推测,25%的变异是有害的。这与SIFT的20%假阳性错误相似,后者表明大多数非同义SNP在功能上是中性的。此外,dbSNP中预测影响功能的变异体子集与证实SIFT敏感性的疾病有关。
SIFT算法仅依赖序列进行预测,但其性能与使用结构的工具类似(三,6–8). 不需要结构的一个优点是可以预测更多的取代。在SNP联盟鉴定的非同义SNP中,74%与蛋白质序列数据库中的同源物足够相似,用于SIFT预测。随着更多基因组测序和更多蛋白质序列可用,SIFT可以预测的替代数量预计将增加。
SIFT预测方法
SIFT假设重要的氨基酸在蛋白质家族中是保守的,因此保守位置的变化往往被预测为有害的。例如,如果蛋白质家族排列中的一个位置仅包含氨基酸异亮氨酸,则假定对任何其他氨基酸的取代是针对的,并且异亮氨酸对于蛋白质功能是必要的。因此,任何其他氨基酸的变化都会对蛋白质功能有害。如果排列中的一个位置包含疏水性氨基酸异亮氨酸、缬氨酸和亮氨酸,那么SIFT实际上假设该位置只能包含具有疏水性的氨基酸。在这种情况下,其他疏水性氨基酸的变化通常可以耐受,但其他残基(如带电或极性)的变化将影响蛋白质功能。
为了预测蛋白质中的氨基酸替换是否会影响蛋白质功能,SIFT考虑了变化发生的位置和氨基酸类型的变化。给定一个蛋白质序列,SIFT选择相关蛋白质并通过查询获得这些蛋白质的比对。根据排列中每个位置出现的氨基酸,SIFT计算了一个位置的氨基酸在最常见的氨基酸被耐受的条件下被耐受的概率。如果此标准化值小于截止值,则预计替换是有害的(2). SIFT算法和软件已在前面介绍(2,三).
SIFT网站
输入
用户可以在http://www.blocks.fhcrc.org/sift/sift.html。在此页面中,有三个提交页面的链接,允许用户不同程度的参与,以控制其预测的质量。
用户只需提交他们的蛋白质序列和氨基酸替代品,就可以将参与程度降到最低。在全自动模式下,SIFT将搜索与查询蛋白同源的蛋白质序列,并根据这些序列计算每个可能氨基酸变化的概率。用户可以从SWISS-PROT、SWISS-POT/TrEMBL或NCBI的非冗余蛋白质数据库中进行选择,以便SIFT进行搜索(4,9).
虽然SIFT可以自动选择序列,但当提供的所有序列都与查询蛋白同源时,可以获得更好的预测结果。这是因为包含同源序列会干扰仅在同源序列中保守的残基的预测。如果用户已经拥有与感兴趣的蛋白质功能相似的序列,则可以直接提交这些序列,并跳过SIFT选择序列的步骤。给定查询蛋白和同源序列,SIFT进行比对。
如果区域错位,SIFT将无法识别保守位置,从而错过潜在的破坏性替换。为了获得最佳预测质量,第三种操作模式允许用户提交自己的校准。
输出
对蛋白质中每个位置的所有20种可能的氨基酸变化进行了预测。还将返回对齐,以便用户可以检查用于预测的序列,并修改它们以便重新提交。此选项还可用于从SIFT在自动模式下生成的对齐输出中删除不确定、错误和未对齐的序列。
对于用户提交的氨基酸替代物,提供了更详细的概要(图。1). 分数是氨基酸变化被容忍的标准化概率。SIFT预测得分低于0.05的替代物是有害的。一些SIFT用户发现分数小于0.1的替代物对检测有害SNP具有更好的敏感性(Cornelia Ulrich,个人通信和10). 定量分数允许用户通过从最低分数到最高分数对氨基酸变化进行排序来确定其优先级。
预测为有害的替代的可信度取决于比对序列的多样性。如果用于预测的序列密切相关,那么许多位置将显示为保守,SIFT将预测影响蛋白质功能的大多数替换。这将导致高假阳性错误,其中功能中性替代被预测为有害的。
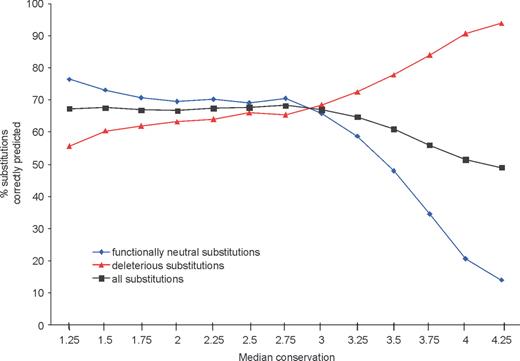
为了提醒用户注意这些情况,SIFT计算中位数保守性值,该值用于测量路线中序列的多样性。以信息含量衡量的保护(11)计算路线中每个位置的,并获得这些值的中值。原木的保护范围220 (= 4.32),当一个位置完全保守并且只观察到一个氨基酸时,当在一个位置观察到所有20个氨基酸时。默认情况下,SIFT使用中值3.0构建路线。基于具有较高中值守恒值的序列比对的预测差异较小,且假阳性误差较大(图。2).
即使可用的同源序列很少,SIFT也比简单地预测非保守氨基酸替代为有害的要好,其中非保守变化被定义为氨基酸替代评分矩阵中的负分数。我们已经表明,只有一个序列与测试蛋白同源,与替代评分矩阵相比,SIFT可以准确预测两倍于中性替代的数量(2). 即使同源序列很少,测试蛋白和其他序列之间的位置也会不同。根据这些位置出现的氨基酸,SIFT可能预测这些位置对蛋白质功能不重要。这些附加信息可以消除功能中性取代,并增加对有害取代的选择性。
总之,可以从诱变项目、SNP数据集和密切相关的生物体之间的变化中获得大量替代。当在所有替换、SIFT和其他类似的预测工具上进行实验不可行时(13)可能有助于确定哪些变化影响蛋白质功能并可能导致表型差异。
致谢
我们感谢Jorja Henikoff的建议和鼓励。这项工作得到了NIH的资助(GM29009)。
图1。SIFT预测蛋白质中氨基酸变化的示例。分数小于0.05的替代物预计会影响蛋白质功能。在最后一次预测中,序列的中值守恒不符合阈值,因此发出警告。
图2。预测取决于比对中使用的序列的多样性。正确预测的替换百分比基于在整个LacI蛋白中分析的4000多个替换大肠杆菌(2,12). 当用于预测的序列序列密切相关(高度中值守恒)时,许多位置显得守恒且对功能很重要。在这种情况下,对有害取代的预测精度很高,但许多功能中性取代被错误地预测为有害。为了获得与特定中值守恒的比对大肠杆菌已提交至SIFT网站,并调整了中值保护设置。因为可用的同源序列与大肠杆菌LacI,无法获得具有较高中值保护值的线形。为了获得中值守恒值大于3.25的比对,从相同比对开始模拟密切相关的序列大肠杆菌LacI序列。从LacI比对中随机选择一个位置和一个序列,中值保持2.75。对应于该位置的氨基酸在起始比对中被取代。氨基酸继续被随机选择和替换,直到达到所需的中位数保守性。然后评估模拟对准的性能,如前所述(2)绘制的值是100条模拟路线的平均性能。
工具书类
1Krawczak,M.、Ball,E.V.、Fenton,I.、Stenson,P.D.、Abeysinghe,S.、Thomas,N.和Cooper,D.N(
2000
)人类基因突变数据库——生物医学信息和研究资源。嗯,变种人。
,15
,45
–51。 2Ng,P.C.和Henikoff,S(
2001
)预测有害氨基酸替代。基因组研究。
,11
,863
–874. 三。Ng,P.C.和Henikoff,S(
2002
)解释预测会影响蛋白质功能的人类多态性。基因组研究。
,12
,436
–446. 4Bairoch,A.和Apweiler,R(
2000
)SWISS-PROT蛋白质序列数据库及其2000年的补充TrEMBL。核酸研究。
,28
,45
–48. 5Sherry,S.T.、Ward,M.H.、Kholodov,M.、Baker,J.、Phan,L.、Smigielski,E.M.和Sirotkin,K(
2001
)dbSNP:NCBI遗传变异数据库。核酸研究。
,29
,308
–311. 6Sunyaev,S.、Ramensky,V.、Koch,I.、Lathe,W.、III、Kondrashov,A.S.和Bork,P(
2001
)有害人类等位基因的预测。嗯,分子遗传学。
,10
,591
–597. 7D.查斯曼和R.M.亚当斯(
2001
)预测非同义单核苷酸多态性的功能后果:基于结构的氨基酸变异评估。分子生物学杂志。
,307
,683
–706. 8桑德斯,C.T.和贝克,D(
2002
)评估结构和进化对有害突变预测的贡献。分子生物学杂志。
,322
,891
–901. 9Wheeler,D.L.,Church,D.M.,Lash,A.E.,Leipe,D.D.,Madden,T.L.,Pontius,J.U.,Schuler,G.D.,Schriml,L.M.,Tatusova,T.A.,Wagner,L.和Rapp,B.A(
2002
)国家生物技术信息中心数据库资源:2002年更新。核酸研究。
,30
,13
–16. 10Leabman,M.K.,Huang,C.C.,DeYoung,J.,Carlson,E.J.,Taylor,T.,de la Cruz,M.,Johns,S.J.,Stryke,D.,Kawamoto,M。,等.(2003)人类膜转运蛋白基因的自然变异揭示了进化和功能限制。程序。美国国家科学院。科学。美国,正在印刷中。
11Schneider,T.D.、Stormo,G.D.、Gold,L.和Ehrenfeucht,A(
1986
)核苷酸序列上结合位点的信息含量。分子生物学杂志。
,188
,415
–431. 12佩斯,H.C.,科尔彻,M.A.,卢,P.,马基维茨,P.、米勒,J.H.,张,G.和刘易斯,M(
1997
)真实空间中的Lac阻遏物基因图。生物化学趋势。科学。
,22
,334
–339. 13拉蒙斯基,V.,博克,P.和苏尼亚耶夫,S(
2002
)人类非同义SNPs:服务器和调查。核酸研究。
,30
,3894
–3900.







{kind=link}

{kind=link}