GROMACS公司 -最广泛使用的HPC应用程序之一,随着 GROMACS 2020年 新版本包括NVIDIA和 核心GROMACS开发人员 .
作为生物分子系统的模拟软件包,GROMACS 使用牛顿运动方程演化粒子。 强制命令 运动:例如,两个带正电的离子相互排斥。 精明的 力是模拟中最昂贵的部分,因为所有成对的 粒子可能会相互作用,模拟涉及许多粒子。
GROMACS提供了广泛的功能 不同类型的力计算。 对于大多数模拟 重要类别(就计算费用而言)包括:
非结合短程力: 在一定截止范围内的粒子被认为是直接相互作用的。 颗粒网格Ewald(PME)远程力: 对于较大的距离,力通过一种方案进行建模,其中傅里叶变换用于在傅里叶空间中进行计算。 这比直接在实际空间中计算所有交互作用要便宜得多。 粘结短程力: 由于粒子之间键的特定行为,例如,拉伸两个共价键原子时的谐波电势,也需要这样做。
在以前的GROMACS版本中,这些力类已经支持GPU加速(PME力计算中使用了CUDA快速傅里叶变换库)。 最新加入的是2019系列中的GPU保税部队,该部队是通过NVIDIA和核心GROMACS开发商之前的合作开发的。
然而,仍然存在一个问题。 力计算有 在现代GPU上速度如此之快,以致于模拟的其他部分 在计算费用方面非常重要,特别是当您想要 对单个模拟使用多个GPU。
本文介绍了2020版本中可用于解决此问题的新性能功能。 对于许多典型的模拟,整个时间步长现在可以在GPU上运行,避免了CPU和PCIe瓶颈。 现在,GPU之间的通信操作可以直接在GPU内存空间之间运行。 这项工作的结果表明,性能有了很大提高。
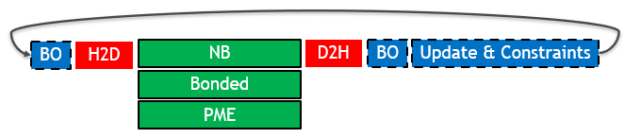
单GPU移植和优化 下图(图1)描述了在单个GPU和之前的2019版本上运行时,典型GROMACS仿真中单个时间步长的关键部分(时间步长从左到右运行):
图1。 使用2019年GROMACS版本的单个时间步长的关键部分。
不同的框样式表示每个零件的硬件分配:
在GPU上执行(带实心边框的绿色框):显示了三种类型的力计算,它们可以相互重叠。 在CPU上执行(带虚线边框的蓝色框):缓冲操作(BO)是内部数据布局转换和强制减少操作。 更新和约束是时间步的最后一部分,其中原子坐标将使用先前计算的力进行更新和约束。 数据传输(无边框的红色框):所需的PCIe主机到设备(H2D)和设备到主机(D2H)数据传输。
通用分组 只加速力的计算是一种有效的策略 体系结构。 即使有了加速度,它们仍然占据主导地位 时间步长的其他部分在性能方面并不重要。 然而, 在现代架构(如Volta)上,力核现在非常快 其他间接费用也变得很大。
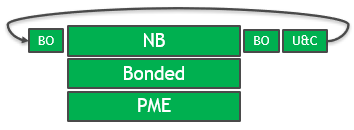
GROMACS 2020版本中的解决方案是所有关键计算部分的完全GPU支持。 GPU在整个时间步长内使用,并且消除了重复的PCIe传输,如图2所示
图2。 使用GROMACS 2020优化时间步长。
PME力内核也进行了优化,以允许更多 有效地将工作分配给CUDA线程和更好的内存访问模式。
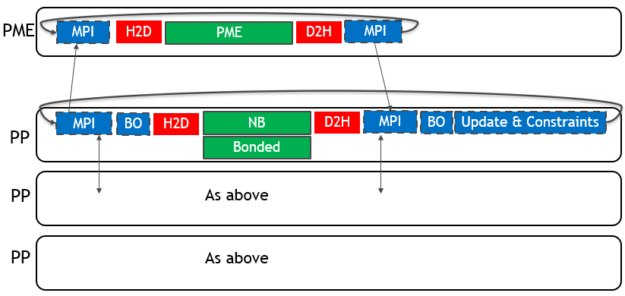
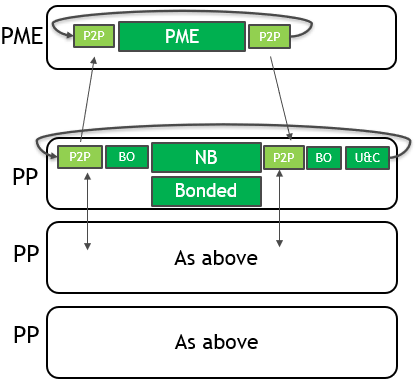
多GPU优化 为了将多个GPU用于单个仿真,GROMACS将PME力计算分配给单个专用GPU。 它分解了剩余GPU上的非键合力和键合力计算。 单个MPI任务将控制每个GPU:这些任务分别称为PME任务和Particle-Particle(PP)任务。 下图(图3)描述了2019年版4-GPU仿真的情况:
图3。 使用GROMACS 2019进行4-GPU仿真的单个时间步长的关键部分。
对于单个GPU情况,只有力计算(在 绿色带实线边框)卸载到GPU,CPU(蓝色带虚线 边界)和PCIe(红色,无边界)组件限制性能。
现在需要两种类型的通信: 具有“光环交换”性质的PP任务之间(其中 将每个域的边缘转移到相邻域) PME和PP任务之间。 对两个坐标数据都执行这些操作 (靠近每个时间步长的开始)和力数据(靠近每一时间步长结束)。
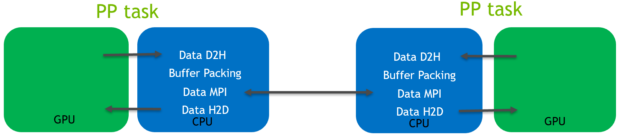
在这个预先存在的代码路径中,数据通过 CPU,如下图(图4)所示,用于光环交换 坐标数据:
图4。 坐标数据halo交换的代码路径,使用GROMACS 2019。
如图4所示:
主机到设备(H2D)和设备到主机(D2H) 在GPU和CPU之间传输数据需要CUDA内存副本 内存空间。 它们使用相对低带宽的PCIe总线。 在CPU上执行缓冲区打包。 是的 必需,因为要传输的数据不连续。 在CPU内存空间上操作的MPI调用 用于在控制独立GPU的MPI任务之间传输数据。
使用GROMACS 2020,这些通信操作现在可以在GPU内存空间之间直接执行,并自动路由,包括使用NVLink(如果可用),如图5所示:
图5。 使用GROMACS 2020优化时间步长,用于4-GPU模拟。
新战略如下图所示 (图6):
图6。 坐标数据halo交换的代码路径,使用GROMACS 2020。
CUDA对等(P2P)内存副本现在可以传输数据 直接在GPU内存空间之间。 它们穿越高带宽NVLink 互连(如果可用)。 即使在没有NVLink的系统上 由于它提供的异步性,该机制的速度大大加快。
这些P2P传输是由以下事实促成的: 缓冲区现在使用CUDA内核直接封装在GPU内存空间中。 这个 需要有关传输中涉及的数组索引的信息,但 信息不会每次都更改。 只有当 分解变化。 您很少在CPU和 传输到GPU,对性能没有显著影响。
GROMACS提供了一个名为“线程MPI”的内部MPI库,它在后台使用CPU线程。 因此,对于主要的P2P数据传输,您可以利用多个MPI任务位于同一进程中这一事实,并使用在统一虚拟地址空间上操作的CUDA内存副本。 然而,这意味着优化仅限于单个计算节点(对于不支持的情况,会自动进行回退)。 目前正在进行进一步的工作,以扩展该机制,消除这一限制。
每个MPI任务都必须知道哪些远程内存位置 写入和读取,还必须使用远程CUDA事件来允许CUDA 以在不同的GPU之间正确同步。 这些是在以下双方之间交换的 MPI任务在更改时使用MPI发送和接收通信。
早期的图表显示了坐标的光晕交换 PP任务之间的数据。 力数据的光晕交换机制 PP任务之间是类似的,其中需要缓冲区解包,而不是 包装。 同样,PP和PME之间的协调和强制通信 任务类似,但缓冲区打包和解包不是必需的数据 是连续的。
为了演示这些好处,我们展示了运行多GPU的结果 选择三个测试用例来代表 真实世界模拟的典型系统尺寸范围。 从最小到 最大的,我们使用了以下测试用例模拟:
多动症(95561个原子) 纤维素(408609个原子) STMV(1066628个原子)
我们使用了四个NVIDIA V100-SXM2 16GB GPU(带有完整的NVLink 连接)由两个Intel Xeon E5-2698 v4(Broadwell)20核CPU托管, 其中(如上所述),我们为PME分配了一个GPU,并将PP分解为 其余三个GPU。
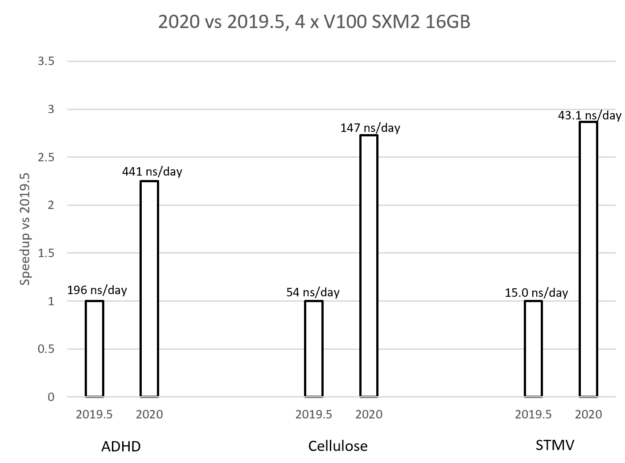
图7。 使用三个多GPU仿真示例比较2019年GROMACS和2020年GROMAC。
图7中的图表比较了新的GROMACS 2020版本(启用了早期优化)与之前的2019.5版本在每个测试用例中的性能。 该图显示,每种情况下实现的加速比都在2X到3X之间,其中加速比随问题大小而增加。 标签显示了每种情况下的实际性能,以纳秒/天为单位,即在运行模拟的一天内可以模拟多少纳秒。 这些性能优势是由于本文中描述的改进的结合。
当在单个GPU上运行时,多GPU通信的改进当然是不相关的,因此改进不那么显著。 然而,由于整个时间步长可以在GPU上运行,并且避免了昂贵的PCIe传输,因此仍然有很大的优势。 将多个仿真分配给同一个GPU时,您可能希望根据使用中的情况使用预先存在的代码路径–使用可用选项进行实验。
在运行上述性能比较时,但仅限于一个GPU(和一个主机CPU),我们观察到,在三个测试用例中,2020版(启用了所有优化)的加速比上一个2019版在1.4X–1.5X的范围内。
为模拟启用优化 此帖子中的优化尚未由启用 违约。 新的代码路径已经过标准GROMACS的验证 回归测试,但仍然缺乏实质性的“真实世界”测试。 我们强烈要求 建议您根据默认路径仔细验证结果。 任何 所报告的问题将被开发团队感激地接收以提供帮助 使软件成熟。
GROMACS应使用其内部 螺纹MPI 库而不是任何外部MPI库。 有关更多信息,请参阅前面的Multi-GPU优化部分。
在运行时,可以通过将以下三个环境变量设置为shell中的任何非NULL值来完全启用优化(此处显示的是bash shell)。
对于PP任务之间的光环交换通信,请使用以下命令:
export GMX_GPU_DD_COMMS=真 对于PME和PP任务之间的通信,请使用以下命令:
导出GMX_GPU_PME_PP_COMMS=真 启用时间步长的更新和约束部分 对于多GPU:
导出GMX_FORCE_UPDATE_DEFAULT_GPU=true 这些设置的组合将触发所有 优化,包括依赖项,如缓冲区的GPU加速 操作。
在单个GPU上运行时,仅 GMX_FORCE_UPDATE_DEFAULT_GPU公司 是必需的(如果仅用于单GPU,则可以通过添加 -更新gpu 选项 mdrun软件 命令)。
在单GPU和多GPU情况下,还需要通过以下选项将三类力计算分配给GPU mdrun软件 :
-nb gpu-键合gpu-pme gpu 在多GPU上 -核动力装置1 选项也需要将PME限制为单个GPU。
当使用更新组时,新的GPU更新和约束代码路径仅支持与跨多个GPU的PP任务的域分解结合使用。 这意味着除了小分子外,不支持约束所有键。
为了促进这一点,我们建议您将氢键的键转换为约束,而不是所有键。 为此,在GROMACS预处理器grompp的.mdp输入文件中,更改以下行:
约束=所有债券 新行应如下所示:
约束=氢键 有关更多信息,请参阅 mdp选项 用户指南的第节。
这是全套 mdrun软件 运行4-GPU性能比较时使用的选项:
gmx mdrun-v-nsteps 100000-resetstep 90000-noconfout\ -ntmpi 4-ntump 10\ -nb gpu-绑定gpu-pme gpu-npme 1\ -nstlist 400 第一行指示GROMACS为这个相对较短的基准测试运行100000个步骤。 定时计数器在步骤90000重置,以避免定时中包含初始化成本,因为这些对于长时间生产运行通常并不重要。 选择这些特定值是为了让GROMACS有足够的时间自动执行PME调整,其中PME网格的大小被调整,以在PP和PME任务之间实现最佳负载平衡。 类似地 -诺康福特 选项指示GROMACS在短期基准测试运行结束时不写入输出文件,以避免人为的高I/O开销。
第二行指定四个thread-MPI任务应该 使用(每个GPU一个),每个线程10个OpenMP线程-MPI任务。 共 正在使用40个OpenMP线程来匹配服务器中物理CPU内核的数量。
第三行将所有力计算卸载到GPU, 如前所述。
决赛 -nstlist 400标准 选项指示GROMACS以400步的频率更新邻居列表。 使用Verlet方案时,可以调整此选项,而不会损失任何精度。 我们通过实验发现该值可以提供最佳性能。
接下来的步骤 有关使用的更多信息,请参阅GROMACS 2020 发行说明 和 用户指南 有关此帖子中描述的发展的更多信息,请参阅GTC 19演示文稿 在现代多GPU系统上加速GROMACS ,可进行录制。