摘要
动机:需要有效的拟合工具来利用结晶学或比较建模中蛋白质结构域原子模型的快速增长,以及较大分子组装体的低分辨率密度图。在这里,我们报告了一种新的拟合算法,用于将部分高分辨率模型完全快速地覆盖到低分辨率密度图中。该方法结合了基于球谐函数(SH)的快速旋转搜索和简单的平移扫描。
结果:这种新颖的组合使得可以在几秒到几分钟的时间内将原子结构精确地对接到低分辨率的电子密度图中。通过模拟和实验测试用例实现的高效性保留了这些异构分辨率合并工具所需的详尽性。结果证明了它的效率、鲁棒性和高吞吐量覆盖率。
可利用性: 作者网页
联系人: pablo@cib.csic.es
补充信息:补充数据可在生物信息学在线。
1简介
刚体拟合是通过可用的原子结构成分解释大分子结构电子显微镜(EM)图中包含的信息的标准方法。这是一个复杂的拼图游戏,其中大分子的低分辨率三维EM密度图充当模糊框架,指导连锁原子分辨率碎片的集合。完成后,这个拼图游戏产生了整个大分子的近原子细节图片。因此,通过解决这一难题,我们可以更好地了解主要细胞过程中中心参与者的内部工作。
在过去几年中,开发了许多高性能拟合算法和程序。EMFIT等项目(罗斯曼,2000),COAN公司(Volkmann和Hanein,2003年),码头(罗斯曼,2000年)、FOLDHUNTER(江等., 2001),颜色(Chacon和Wriggers,2002年)SITUS的(Chacon和Wriggers,2002年;箭牌手表等., 1999)财务风险管理(科瓦奇等., 2003)、URO(纳瓦扎等., 2002)和3SOM(Ceulemans和Russell,2004年)已成功用于提供有关大分子功能的结构见解。本质上,这些工具对所有可能的相对旋转和平移执行自动搜索,以最大化密度相关函数。这种相关性通常在目标实验EM图和通过降低待对接原子结构的分辨率获得的模拟探针图之间进行计算[有关审查,请参阅Wriggers和Chacon,2001年和Fabiola和Chapman,2005年]. 尽管成功应用,但大多数这些对接工具执行的彻底搜索是一个非常耗时的过程,因此它们还不足以支持高通量的装配过程。
随着当前结构基因组学的努力,结构建模的进展以及天然环境中大分子3D成像的前景(Baumeister和Steven,2000年;卢奇克等., 2005),需要开发更快的算法(尼科尔等., 2006;罗素等., 2004;萨利等., 2003).
首选的拟合算法应平衡效率和稳健性,其中效率通常与降低的计算成本相关,稳健性与对接搜索的准确性和彻底性相关。多分辨率结构谜题可能非常复杂,可能会由于一些复杂因素导致不同的对接姿势:分辨率差异、EM图的低信噪比、原子结构和EM结构之间的偏差(例如缺失区域、无序和构象变化)等。高速算法可以提供独特的关键优势。它将允许扫描大量可能的替代模型,以便将域安装到更大的密度图中。例如,通过EM成像的分子的单个组分的原子结构未知是很常见的。在这种情况下,可以求助于同源性建模,这可以为我们提供一套广泛的潜在原子模型。Topf和合作者使用MODELLER(Fiser和Sali,2003年)生成可放置在综合体目标3D EM图中的替代比较模型(顶部(Topf)等., 2005;Topf和Sali,2005年). 这些作者还证明了使用中分辨率EM图来提高比较建模准确性的逆任务的有用性(顶部(Topf)等., 2006). 如果相关结构不可用,当地图分辨率高于12º时,可以应用折叠分配和模板选择程序。SPI-EM公司(贝拉兹·穆里尔等., 2005)使用统计方法和对接搜索相结合的方法,能够确定哪些CATH超家族域可以对接到目标EM图中。相关对接程序,如Helixhunter(江等., 2001)或EMatch(德罗尔等., 2007)包括模板匹配程序,以识别3DEM地图中的二级结构元素。所有这些方法都将受益于可以执行高效刚性查询搜索的新方法。
在这里,我们提出了一种新的多分辨率对接方法,该方法在效率和精度之间取得了很好的平衡。该方法是快速旋转匹配(FRM)方法的一种新组合(科瓦茨和箭手,2002年)使用平移扫描,也可以被视为FRM5D方法的实际简化,如科瓦奇等., 2003。与FRM5D中涉及五个角度和一个平移参数的公式不同,这里我们只使用FRM加速三个旋转自由度(DOF),而只扫描三个平移自由度。通过球谐函数(SH)和一个方便的3D旋转群公式以及优化的代码设计,我们能够实现搜索旋转空间的高效性和穷尽性。这种新方法不受FRM5D方法的强大内存限制的影响(科瓦奇等., 2003)并构成了一个快速、健壮的多查询对接搜索工具。各种模拟和实验三维EM图的结果证实了其有效性和适用性。此外,所开发的方法是一种通用的注册工具,可以轻松应用于任何3D刚体注册问题。
2方法
搜索问题的计算解决方案可以简化为找到相对方向和平移,从而最大化待停靠结构/地图的密度互相关。在这种情况下,对于给定的旋转和平移,拟合标准通常定义为EM实验图ρ之间的标量积低的和原子结构的低通滤波版本ρ高的,数学上:其中Ω吨和∧对分别表示平移算子和旋转算子。为了找到最高相关值,以前的方法将对探针结构(通常为ρ高的)相对于固定基准(ρ低的)将其与基于卷积定理的快速傅里叶变换(FFT)加速平移搜索相结合。这个众所周知的详尽搜索协议是从蛋白质-蛋白质对接领域借用的(加布等., 1997;卡查尔斯基-卡齐尔等., 1992;瓦克瑟等。, 1999),除其他外,由事实上的标准多分辨率对接工具COLORES(Chacon和Wriggers,2002年). 作为通过FFT加速平移自由度的替代方法,我们加快了旋转搜索,从而如本文所示,提供了更高的效率。该方法称为FRM,使用SH对3D旋转组进行适当的参数化,以有效计算相关函数的旋转部分。其他地方对FRM方法的理论进行了详细描述(科瓦奇等., 2003;科瓦茨和箭手,2002年). 简单地说,要对接的密度函数首先由SH函数中的展开式近似。为此,密度体积被划分为同心球面层(如洋葱壳),每个球面层由有限和近似表示为:哪里:而不是将穷尽的搜索重铸为一个包含五个角度和一个平移参数的公式(科瓦奇等., 2003),这里我们只对三个旋转自由度进行加速,而对三个平移自由度进行简单扫描。仅考虑旋转部分,拟合函数现在可以用SH变换的傅里叶逆变换表示[方程式(1)]密度图的(科瓦茨和箭手,2002年):其中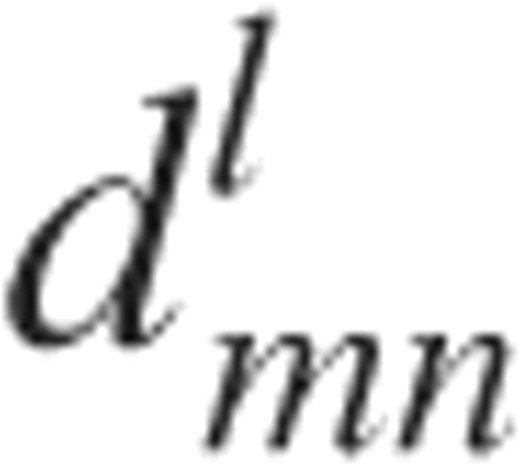 是定义3D旋转组不可约表示的矩阵元素的实系数。通过预计算系数,可以非常有效地计算此表达式并将密度非零值的最大壳半径用作积分上限。通过这种方式,方程式(2)对于给定的平移,可以非常快速地计算所有旋转的相关函数,其采样频率为带宽的两倍B类用于地图的调和变换[公式(1)]. 例如,B类=16对应于扫描~16 000圈,采样步长为11.25°。如果旋转采样步长设置为5.6°(B类= 32), >130 将探索1000次旋转。因此,该方法提供了一种适应性强的精细旋转筛选。
是定义3D旋转组不可约表示的矩阵元素的实系数。通过预计算系数,可以非常有效地计算此表达式并将密度非零值的最大壳半径用作积分上限。通过这种方式,方程式(2)对于给定的平移,可以非常快速地计算所有旋转的相关函数,其采样频率为带宽的两倍B类用于地图的调和变换[公式(1)]. 例如,B类=16对应于扫描~16 000圈,采样步长为11.25°。如果旋转采样步长设置为5.6°(B类= 32), >130 将探索1000次旋转。因此,该方法提供了一种适应性强的精细旋转筛选。 C类 勒姆 (第页)是与特定的复值球谐函数相关的系数Y(Y)勒姆(β, λ) 在单位球体上定义。
我≥0和−我≤米≤我是SH度和顺序,β和λ分别是同纬度和经度。
根据采样定理,使用的采样点数量(在每个β和λ中)等于带宽的两倍B类。
然后通过对均匀覆盖平移搜索空间的采样点列表应用此FRM旋转扫描来执行穷举搜索。为了防止探索没有物理意义的点,平移空间被限制在探针尺寸(原子结构)大致符合实验EM图的位置。为此,遮罩由目标地图内的点定义,并被探针结构的最小半径侵蚀。已经实施了替代的(和更有效的)平移搜索策略,例如径向搜索(适用于带孔结构)或基于中心的搜索(适用于带相似尺寸的对接结构)(科瓦奇等., 2003). 这些采样方案利用了几何学的优点,但其应用范围并不像使用掩模的均匀采样方案那样具有普遍性。因此,这里我们只报告使用掩蔽策略获得的结果。虽然可以使用较大的平移采样,但在我们的测试中,我们发现通过探索其他每个体素,我们没有遗漏任何显著的相关峰值。这是可能的,因为相关性是使用六个3D相邻位置之间的简单抛物线近似插值的。选择平移采样是一种在穷尽性和效率之间折衷的实用解决方案。还将进行进一步的工作,以建立更大的采样限制,并在其中授予穷尽性,或开发新的高效粗网格搜索策略。
密度互相关工作得相当好,尽管在特定情况下,将其用作对接标准可能会导致不明确的匹配或误报。当将小组件放置在大密度图中时,这在低分辨率(低于15º)下可能非常关键。可以采用几种替代方案来提高拟合对比度。例如,可以根据局部相关准则进行拟合(Rath公司等., 2003;罗斯曼,2000),或者可以使用拉普拉斯核对映射进行预过滤(Chacon和Wriggers,2002年). 由于它的实现不需要对注册方案进行任何更改,因此这里使用拉普拉斯滤波贴图而不是原始密度贴图执行部分对接。用拉普拉斯核卷积映射的策略通过同时包含密度和轮廓重叠来改善潜在解之间的数值对比。尽管其已知的局限性,例如对高频噪声的敏感性以及探头结构的表面暴露相对有限的情况,但仍有许多成功的应用报道;参见示例(戈拉斯等2003年;劳林梅基等., 2005;莱曼等。, 2004;欧帕尔卡等。, 2003;佩托萨等。, 2004;萨姆索等., 2006;桑丁等., 2004;苏厄尔等., 2003).
为了将多分辨率对接技术扩展到更高的吞吐量覆盖范围,对FRM和平移扫描的新组合的实现进行了仔细设计和优化,以实现最大的运行时节省。这种新算法称为ADP_EM(EM的另一个对接平台),是用C++编写的,以获得面向对象方法的灵活性和可重用性。
3结果
3.1对接基准
我们的新型对接算法的性能首先在28个模拟对接案例上进行了测试,这些案例包括多种高分子形状(参见图1详细列表)。每个测试用例包括五个模拟的3D-EM图,实验分辨率为10、15、20、25和30º,以及用于生成此类密度图的大分子结构的原子亚单位或组分。通过执行对接过程,每个测试用例的原子子单元应该正确定位到相应的完整EM图中。因此,使用该基准,我们评估了我们的方法在最具挑战性的情况下的性能,即待对接的原子结构仅代表低分辨率密度图的一部分。为了具有统计意义并避免预对齐情况,对于每个原子成分,从不同的相对位置开始重复50次注册搜索。此外,使用了三种不同的旋转采样:~11°、8°和6°,分别对应于16、24和32的谐波带宽。(请参见图1获取所用参数的完整描述。)
图1
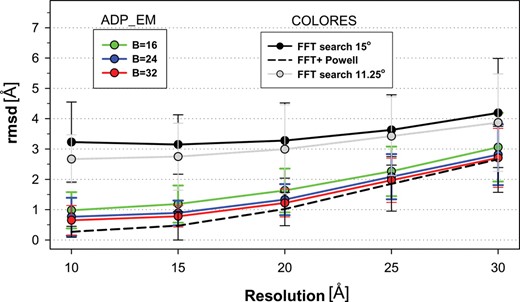
注册准确性。利用EMAN软件包的PDB2MRC程序降低28个原子大分子结构的分辨率,在模拟EM图上进行对接测试(卢特克等., 1999). 所选分辨率(10、15、20、25和30º)对应于EM测量的典型实验范围。所用的测试结构为:5-氨基戊二酸脱水酶(PDB:1aw5,8×对称相关拷贝);甲基辅酶M还原酶(1e6v)的A链(2×)、B链(2 x)和BC链(2倍);谷氨酰胺合成酶(1fpy,10×);ATP硫酰化酶(1g8g,6×);三角蛋白酶为同六聚体(1k32,6×);喹啉-呋喃酸还原酶(1kf6)的A链(2×)和CD链(2 x);ATP硫酰化酶(1g8g,6×),全甘油醛-3-磷酸脱氢酶(1gd1,4×);谷氨酸脱氢酶(1l1f,6×);作为同源三聚体的三角蛋白蛋白酶(1n6d,3×);亚硝酸铜还原酶(1nic,3×);蛋白酶体α环(1j2p,7×);钙粘蛋白(1q5b,3×);凝集素(1w3a,6×);血凝素(1ruz,3×)RecA(1xmv,6×);电压门控钾通道β2-亚单位链A(2a79,4×);菌毛(2菌毛,5×);过氧化氢酶(7cat,4×);GroEL ATP(7×)和ADP亚基(7×;保温箱(1a6d,14×);核糖体大小亚基(1ffk+1fjf);和核糖体蛋白S2(1ffk−1fjf)。为了增强统计并防止每个测试结构出现预对齐情况,从原子结构的不同平移和旋转副本开始重复注册搜索50次。使用三个不同的旋转采样步骤对所有测试用例进行注册程序:11°、8°和~6°,分别对应16、24和32的谐波带宽。在所有情况下,平移采样被选择为6º,这是地图网格大小的两倍。为了验证目的,计算了最高相关拟合亚基和原始大分子中包含的等效结构(用于生成模拟图)之间的全原子rmsd。为了测量配准精度,已考虑了所有情况,但方法失败的情况除外。少数失败病例的1aw5、2pil、1e6v和核糖体蛋白S2分辨率为30°,而GroES分辨率低于15°。为了执行FFT平移搜索,COLORES与“–nopowell”选项一起使用,旋转采样步长为15°和11.25°(后者对应于B=16)(分别为实心黑色和灰色线),并使用所有其他参数的默认值。所示结果表示从不同相对位置开始的10次跑步的平均值。通过鲍威尔最小化(黑线虚线)在15°下基于傅里叶变换的COLORES搜索中获得的最佳拟合结果,获得了子体素精度。
在这一彻底的验证测试中获得的结果表明,即使在低分辨率下,该算法也能够以合理的精度为21个目标中的绝大多数目标找到正确的位置 执行了000次对接搜索。在图1最佳对接结果与原始目标结构之间的rmsd显示为分辨率和所用带宽的函数。正如预期的那样,随着地图分辨率的降低,对接精度逐渐降低。10º分辨率下的rmsd小于1º,可以认为是完美匹配。20º时,rmsd小于2º,30º时仍接近3º。只有在非常低的分辨率下,才不可能为所有测试用例获得唯一可靠的对接结果。30℃时,与5-氨基乙酰丙酸脱水酶、菌毛、甲基辅酶M还原酶和核糖体蛋白S2对接失败。另一个无法在得分最高的解决方案中找到正确姿势的案例是分辨率低于15Å的GroES。其他作者已经认定GroES是一个非常困难的对接案例(Ceulemans和Russell,2004年;罗斯曼,2000). 在这种情况下,相对较小的尺寸和低分辨率阻碍了可靠对接。
从经验上讲,通过模拟和实验地图可以获得的最大精度始终是地图标称分辨率的1/10(Chacon和Wriggers,2002年). 其他作者将EM实验地图可达到的最大精度扩展到地图分辨率的4/10(Fabiola和Chapman,2005年).
从中可以看出图1,即使使用最小的带宽,ADP_EM的精度也低于这些限制。当带宽从16增加到24,而从24增加到32时,对接精度也有所提高。这些改进在逻辑上是由于更精细的旋转采样和更准确的谐波描述。然而,最佳情况下的最大增益只有0.4º,考虑到这种混合多分辨率对接方法的最大精确度,这几乎可以忽略不计。因此,我们认为16的带宽足以在大多数对接场景中识别正确的姿势。只有当需要额外的旋转精度时,才应考虑更高的谐波阶数。
在表1,对接搜索的计时显示为分辨率和带宽的函数。可以看出,对分辨率的依赖性很小,但谐波阶数的增加大大增加了对接时间。对于B=32,执行对接穷举搜索的平均时间接近222秒,使用B=24和B=16时分别降至113和34秒。小型地图的搜索时间范围为8秒(例如,1nic,40秒三体素)3分钟,用于大核糖体图(100三体素)。这些计时结果表明,这种新的对接方法实现了高效率。
| . | 取样. | 分辨率. |
|---|
| . | B/°. | 10Å. | 15Å. | 20Å. | 25Å. | 30Å. |
|---|
| ADP_EM公司 | 16/11° | 28 | 31 | 35 | 34 | 38 |
| 24/8° | 100 | 108 | 119 | 118 | 123 |
| 32/6° | 226 | 220 | 225 | 216 | 221 |
| FFT搜索 | −/15° | 1697 | 1926 | 2341 | 5028 | 6681 |
| 最小鲍威尔 | −/15° | 375 | 918 | 1747 | 3739 | 6597 |
| . | 取样. | 分辨率. |
|---|
| . | B/°. | 10Å. | 15Å. | 20Å. | 25Å. | 30Å. |
|---|
| ADP_EM公司 | 16/11° | 28 | 31 | 35 | 34 | 38 |
| 24/8° | 100 | 108 | 119 | 118 | 123 |
| 32/6° | 226 | 220 | 225 | 216 | 221 |
| FFT搜索 | −/15° | 1697 | 1926 | 2341 | 5028 | 6681 |
| 鲍威尔极小值 | −/15° | 375 | 918 | 1747 | 3739 | 6597 |
| . | 取样. | 分辨率. |
|---|
| . | B/°. | 10Å. | 15Å. | 20Å. | 25Å. | 30Å. |
|---|
| ADP_EM公司 | 16/11° | 28 | 31 | 35 | 34 | 38 |
| 24/8° | 100 | 108 | 119 | 118 | 123 |
| 32/6° | 226 | 220 | 225 | 216 | 221 |
| FFT搜索 | −/15° | 1697 | 1926 | 2341 | 5028 | 6681 |
| 最小鲍威尔 | −/15° | 375 | 918 | 1747 | 3739 | 6597 |
| . | 取样. | 分辨率. |
|---|
| . | B/°. | 10Å. | 15Å. | 20Å. | 25Å. | 30Å. |
|---|
| ADP_EM公司 | 16/11° | 28 | 31 | 35 | 34 | 38 |
| 24/8° | 100 | 108 | 119 | 118 | 123 |
| 32/6° | 226 | 220 | 225 | 216 | 221 |
| FFT搜索 | −/15° | 1697 | 1926 | 2341 | 5028 | 6681 |
| 最小鲍威尔 | −/15° | 375 | 918 | 1747 | 3739 | 6597 |
据我们所知,这是在任何多分辨率对接工具上执行的最完整的验证测试。此外,对于这种方法和其他方法的未来发展,对接基准已经在线提供。
3.2比较结果
其中最受欢迎的对接程序是Situs(箭牌手表等., 1999)及其基于相关性的详尽搜索工具COLORES(Chacon和Wriggers,2002年). 通过FFT,这是最快的互相关最大化方法。尽管由于加速平移或旋转搜索的相反哲学,很难与ADP_EM进行真正公平的比较,但这里提出的方法显然在效率上具有很大优势。我们使用15°的旋转采样步骤将COLORES应用于上一节中描述的相同对接基准。注意,在ADP_EM(11.25°)中使用更精细的旋转采样将大大增加待探索的旋转次数(从4416次增加到10次 496)以及计算时间。然而,15°采样足以获得与我们的方法类似的总体拟合精度。事实上,大多数基准案例的拟合都是正确的,而失败案例与ADP_EM获得的结果相同。
对于所有基准测试用例,使用COLORES执行FFT平移搜索所需的平均时间为25分钟(对于子体素求精,不使用鲍威尔最小化),而使用我们的方法,当B=16(~11°)时仅为34秒,当B=32(~6°)时,仅为4分钟。此外,我们的方法可以更好地适应规模。随着分辨率逐渐降低,密度分布在更大的体积上。因此,在我们的基准中,随着分辨率的降低,我们有更大的地图。在表1,我们可以观察到FFT搜索的定时是如何由于这种影响而随着分辨率逐渐增加的。APD_EM没有观察到这种行为,因此证明了其显著更好的缩放性能。
最重要的是,我们的方法产生了更好的rmsd值。FFT平移搜索受到体素大小的限制,最佳拟合距离正确解>3°(图1,实心黑线)。相比之下,我们的FRM搜索实现了更高的精度,特别是在更高的分辨率下(图1,彩色线条)。通过优化最佳命中率可以额外提高精确度。默认情况下,COLORES使用鲍威尔最小化步骤来实现最高精度(图1,黑色虚线),但具有显著的额外计算成本(表1). 这些非常低的rmsd值在大多数实际问题中可能没有意义,因为EM图和探针结构之间的不一致(小的缺失或无序区域、小的构象变化等)总是会妨碍完美匹配。细化是COLORES中克服体素大小所施加的平移限制的必要步骤。相反,ADP_EM不需要进一步细化,因为即使在低谐波B阶下,它也能获得合理的rmsd值,该值低于多分辨率对接的经验极限。只有在高分辨率的特定情况下,我们才应该考虑局部最小化,因为在这种情况下,贴图和底层原子结构之间有很好的对应关系。
据我们所知,3DSOM是将原子结构拟合到低分辨率EM图中的最快选择(Ceulemans和Russell,2004年). 这种基于曲面重叠最大化的方法给出了许多解,因此通常很难区分正确解和错误解。事实上,有必要目视检查大量最佳评分解决方案及其变体,以找到与正确答案相对接近的拟合。因此,我们无法使用我们的基准进行系统测试。3DSOM的作者已经指出了这一局限性,以及正确姿势的rmsd可能略高于其他方法获得的rmsds的事实(Ceulemans和Russell,2004年). 无论如何,尽管这种方法对于小地图更快(对于最小的地图为5秒),但ADP_EM随着地图大小而缩放得更好。ADP_EM需要3分钟将大的亚单位对接到整个核糖体图中,而3DSOM几乎需要一个小时。
我们将重点与事实上的标准对接程序COLORES和最快的替代3DSOM,因为其他程序效率较低。同样,我们不考虑FRM5D方法(科瓦奇等., 2003)因为它有很强的内存限制。例如,带宽为B=32需要4.5 GB的内存,这显然使其在当前工作站中的使用更加复杂。然而,FRM5D的预期性能(使用B=16的整个基准测试平均约12分钟)明显低于ADP_EM。
3.3使用实验图进行测试
与模拟基准相比,很少有“金标准”的X射线/电磁实验测试案例可用于验证新方法。这里我们展示了之前用于测试其他方法或由其他实验室提供的五张EM图的结果。第一种情况是大肠杆菌GroEL–23°处的GroES复合物[高分子结构数据库(EMD)ID 1046]。从PDB条目1AON中提取的GroEL原子亚基被正确地安装到整个伴侣蛋白系统的相应七倍体双环中(图2A). 顺反环的X射线结构与相应的重建结构之间的rmsd差异小于1.3º。然而,就像模拟地图一样,GroES子单元的对接失败了。该亚基的低分辨率和相对非常小的尺寸可能是这种失配的原因。另一个14.9Å的GroEL ATP图谱(EMD ID 1047)从其亚基成功重建(数据未显示)。在这种情况下,七元环的对接结构和原始结构之间的rmsd差异为1.1º。
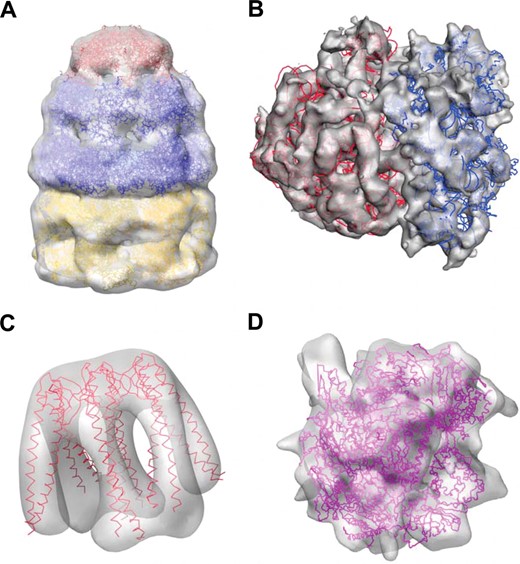
图2
将结果与实验EM数据对接。(A类)大肠杆菌GroES-ADP7-GroEL-ATP7来自大肠杆菌23.5℃(EMD ID 1046,PDB:1ml5);ADP和ATP-GroEL亚基已独立对接,以重建复合体的顺式和反式七元环。对于GroES,使用了整个七肽。(B类)将30S和50S亚单位对接到大肠杆菌14℃的核糖体图谱(EMD ID 1046,PDB:1gix/1giy)。预折叠蛋白的单分子对接(C类)23时(马丁·贝尼托等., 2002)PDB:1l6h,酵母RNA聚合酶II(D类)15度(克雷格黑德等., 2002),PDB:1fxk。
即使原子结构和EM重建之间没有精确的对应关系,我们也获得了很好的拟合。例如,大核糖体亚基和小核糖体子基分别正确地对接到14?图中(EMD条目1005,图2B). prefoldin(蓝丝带)的整体“水母”原子结构很好地符合EM密度,除了已知柔性触手的位置存在微小差异(图2C). 我们还将真核生物RNAPII图与其晶体结构对接詹氏甲烷球菌同系物(图2D). 获得的结果重现了繁琐的手工拟合,该拟合提供了新生RNA的模型,从而提出了RNAPII如何与启动子DNA相互作用的假设(阿斯图里亚斯,2004).
与模拟案例一样,我们能够获得与基于FFT的搜索工具COLORES相同的正确拟合结果(rmsd偏差低于标称分辨率的1/10),但效率更高。例如,使用ADP_EM,将GROEL亚基的任何七元环重建为GROEL-GroES图需要40秒,而COLORES在旋转采样15°的情况下花费了将近1小时(FFT为30分钟,Powell最小化为20分钟)。这种差异随着图谱的扩大而扩大:我们的方法需要296秒才能对接大核糖体亚基,而FFT方法需要近11小时。至于3SOM方法,GroEL-GroES、GroEL-ATP、30S、50S、RNAP和前折叠蛋白的注册分别需要39秒、1米37秒、22米、64米、7米和13米。我们的方法比较快,给出了1.0、1.0、6、13、21330的加速度比。速度的提高是由于ADP_EM具有更好的缩放性能,但在预折叠情况下,加速主要是由于其空心结构,这简化了平移搜索掩蔽策略。在3SOM产生的大量可能的对接姿势中,确定正确的解决方案也存在问题。很难找到一些不同的七倍体GroEL的正确位置,以及核糖体大亚基或RNAP的正确位置。在这些情况下,要么正确的姿势隐藏在次要极小值中,要么其相对于颜色的rmsd和ADP-EM解决方案较高。
3.4同调建模应用
通常情况下,要停靠的组件的原始原子结构未知。在这种情况下,同源建模生物信息学工具提供了一系列潜在有用的原子模型。选择密度相关性最高的模型将很可能导致电子显微镜成像的目标大分子的原子特征。研究表明,与同系物的实验确定的结构相比,比较建模提供的结构在EM图拟合中更有用(顶部(Topf)等., 2005). 事实证明,这种对接过程在比较建模中也可以作为模型评估分数(顶部(Topf)等., 2006).
在这里,我们将ADP_EM对接工具应用于这些作者提供的基准(Topf等。, 2005,作者网页). 基准由八对已知结构的蛋白质组成(每对由一个目标结构及其相应的远程同源物组成,用作建模模板),共享12%到32%的序列一致性。对于每对,使用MODELLER建立了300个备选比较模型(Fiser和Sali,2003年). 基准测试包括从不同分辨率的本地目标结构创建的几个模拟映射。该测试包括通过将所有替代同源模型拟合到相应的密度图中来确定最准确的模型。为了评估模型的几何精度,我们使用MAMMOTH程序对每个靶与其同源物及其相应的比较模型进行了结构比对(奥尔蒂斯等., 2002).
所有模型以及目标和模板原子结构都已使用ADP_EM对接到目标密度图中图3,使用蛋白质1MUP的12°分辨率图绘制拟合相关值与模型比对得分。可以观察到,拟合和模型精度值之间存在明显的相关性。最佳拟合模型总是与结构上接近目标结构的模型相对应。值得注意的是,与模板同系物相比,更接近目标结构的模型通常比同系物本身更好地对接到映射中。不出所料,1MUP目标结构的得分最高,这在逻辑上与完美匹配相对应(见补充表1)。模板、最佳模型和最佳拟合模型的形状非常相似(图3).
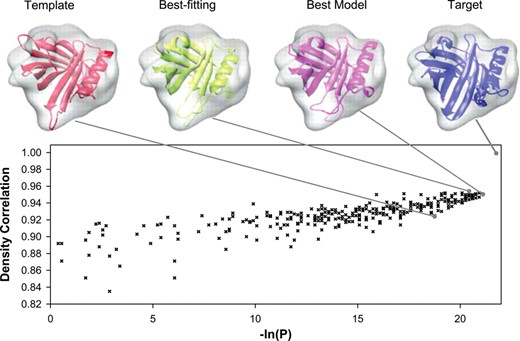
图3
ADP_EM拟合结果,使用中描述的比较同源建模对接基准的1MUP测试用例获得(顶部(Topf)等., 2005)分辨率为12º。目标和模板结构以及300个原子模型已使用ADP_EM对接,旋转精度为11°。根据本地目标结构和使用的300个比较模型之间的MAMMOTH对齐分数,绘制每个模型对接时获得的最佳拟合姿势的密度相关性。对齐分数被定义为−ln(P),其中P是偶然获得相对于最短模型的对齐残数的给定比例的概率(奥尔蒂斯等., 2002). 指向它们的相应值,还显示了模板结构(1rbp,红丝带)、最佳拟合结构(绿色)和最准确模型(粉红色)的最佳拟合。与其他两个更精确的模型相比,模板结构的密度相关性更低。后两者之间的拟合差异几乎不显著。事实上,最佳拟合结构和最佳模型结构彼此非常相似,只能观察到回路和螺旋末端的微小差异。请注意,这是一项严格的测试,因为所有这些结构的形状都非常相似。
然而,对接过程能够区分模型,特别是相对于其他两个模型的模板模型,这两个模型相对于1MUP具有更好的结构重叠。最佳模型在最佳相关性列表中排名第一(6–9)。相反,同源模板结构的比对分数和相关值显著较低(148-167位)。该方法具有鲁棒性,并且与分辨率无关,可获得相同的最佳拟合模型。在所有其他基准案例中也观察到拟合和建模得分之间的良好对应关系,以及为更准确的模型而不是模板获得更好拟合的事实(见补充材料)。这一事实证实了比较建模作为对接协议的潜在用途,正如Toft及其合作者就Mod_EM协议所指出的那样。在这里,我们用一个更高效、更健壮的协议重现了他们的结果。事实上,执行所有302配件的时间为~50分钟,即每次安装10秒。托夫特等.比较改进的FOLDHUNTER方法(江等., 2001)使用优化的扫描蒙特卡罗协议Mod_EM,每次拟合需要约10–15分钟。在拟合方面,所有方法都产生了相当类似的结果。但就效率而言,ADP_EM比这些最具可比性的协议(基于Fourier的穷举搜索协议FOLDHUNTER)至少快60倍。尽管在这个特定的基准测试中,随机蒙特卡罗方法仍然具有竞争力,但在现实世界中,地图比探测器大,预计MC的效率甚至低于FOLDHUNTER(顶部(Topf)等., 2005).
4讨论
ADP_EM比现有方法有了实际的进步,因为它为将X射线晶体结构拟合到低分辨率密度图中提供了一种快速可靠的工具。这种新方法将标准PC上的对接时间减少到几分钟甚至几秒钟。通过模拟和实验测试用例实现的高效性保留了这些异构分辨率合并工具所需的穷尽性。除了节省时间外,我们的方法的主要优点是精细的旋转采样步骤(11到6度之间),可以用于对接搜索,同时仍然保持较高的效率。这确保了全面的6D探索,避免忽略可能的有效对接方案。
达到的性能水平克服了以前的近似值,打开了一个新的应用程序窗口,需要快速而健壮的6D穷举搜索。对于给定的低分辨率结构,通常的做法是使用多个不同的探针进行多次对接,或者解决缩放不确定性。这将有效地有助于获得大分子复合物的精确近原子解释。此外,这种新方法大大简化了来自不同结构源(包括生物信息学建模)的三维信息数据的大规模合并。在此背景下,我们报告了一个同源建模测试用例,作为一个改进的优化协议的示例,该优化协议用于将比较建模结构拟合到EM重建中。这个例子可以很容易地进行缩放,以支持不同方法的大量比较模型(Ginalski,2006年;特雷斯等., 2005),包括使用自动化web服务器(费舍尔,2006年). 还有其他需要高吞吐量覆盖的场景。最吸引人的是用于低温电子层析成像的模板匹配方法(尼科尔等。, 2006)或用于将CATH超家族定位到3D-EM重建中的混合方法(贝拉兹·穆里尔等。, 2005). 后者最近通过利用超家族的结构变异性进行了扩展,以实现柔性装配(贝拉兹·穆里尔等。, 2006). 此外,ADP_EM将是一个非常有趣的工具,用于扫描模型的多个基于灵活性的变体,例如使用低分辨率模型的相关正常模式生成的变体。所有这些有希望的策略都基于广泛的模型拟合步骤,这些步骤可以从我们的超快速可靠对接工具中获得巨大的收益。
由于该方法构成了一种高效的通用三维配准算法,其应用范围可以扩展到其他领域。我们目前正在将所提出的算法应用于蛋白质-蛋白质对接。
作者想感谢J.M.Valpuesta【西班牙CNB(CSIC)】为Prefoldin和F.Asturias(美国斯克里普斯研究所)重建RNAPII。这项工作得到了MEC BFU2004-01282/BMC和BBVA基金会(P.C.)以及NIH 1-R01-GM071872-01(R.A.)的资助。
利益冲突:没有声明。
参考文献
. RNA聚合酶II结构和起始前复合物的组织
, 货币。操作。结构。生物。
, 2004
,卷。 14
(第121
-129
) , . 结构基因组时代的大分子电子显微镜
, 生物化学趋势。科学。
, 2000
,卷。 25
(第624
-631
) , . 用表面重叠最大化法快速拟合低分辨率电子密度图中的原子结构
, 分子生物学杂志。
, 2004
,卷。 338
(第783
-793
) , . 基于多分辨率轮廓的高分子结构拟合
, 分子生物学杂志。
, 2002
,卷。 317
(第375
-384
) 等溶液中酵母RNA聚合酶II的结构:酶调节和与启动子DNA相互作用的意义
, 结构(拱度)
, 2002
,卷。 10
(第1117
-1125
) 等EMatch:一种将原子分辨率亚单位排列成大分子的中分辨率冷冻EM图的有效方法
, 《水晶学报》。生物学博士。结晶器。
, 2007
,卷。 63
(第42
-49
) , . 高分辨率结构与电子显微镜重建图像的拟合
, 结构(拱度)
, 2005
,卷。 13
(第389
-400
) . 蛋白质结构预测服务器
, 货币。操作。结构。生物。
, 2006
,卷。 16
(第178
-182
) , . Modeller:基于同源性的蛋白质结构模型的生成和细化
, 方法。酶制剂。
, 2003
,卷。 374
(第461
-491
) 等利用形状互补、静电和生物化学信息模拟蛋白质对接
, 分子生物学杂志。
, 1997
,卷。 272
(第106
-120
) . 蛋白质结构预测的比较建模
, 货币。操作。结构。生物。
, 2006
,卷。 16
(第172
-177
) 等多蛋白剪接因子SF3b的分子结构
, 科学类
, 2003
,卷。 300
(第980
-984
) 等弥合信息鸿沟:中分辨率结构解释的计算工具
, 分子生物学杂志。
, 2001
,卷。 308
(第1033
-1044
) 等分子表面识别:用相关技术测定蛋白质及其配体之间的几何拟合
, 程序。美国国家科学院。科学。美国
, 1992
,卷。 89
(第2195
-2199
) 等基于五自由度快速傅里叶变换加速度的刚体快速旋转匹配
, 《水晶学报》。生物学博士。结晶器。
, 2003
,卷。 59
(第1371
-1376
) , . 快速旋转匹配
, 《水晶学报》。生物学博士。结晶器。
, 2002
,卷。 58
(第1282
-1286
) 等膜蛋白调节细菌病毒Bam35的双层曲率
, 结构
, 2005
,卷。 13
(第1819
-1828
) 等T4噬菌体感染宿主后尾部蛋白质的三维重排
, 单元格
, 2004
,卷。 118
(第419
-429
) 等电子断层扫描的结构研究:从细胞到分子
, 每年。生物化学评论。
, 2005
,卷。 74
(第833
-865
) 等EMAN:用于高分辨率单粒子重建的半自动软件
, J.结构。生物。
, 1999
,卷。 128
(第82
-97
) 等真核预折叠蛋白及其与未折叠肌动蛋白和细胞溶质伴侣蛋白CCT复合物的结构
, EMBO J。
, 2002
,卷。 21
(第6377
-6386
) 等关于EM重建中模型电子密度的拟合:一个倒数空间公式
, 《水晶学报》。生物学博士。结晶器。
, 2002
,卷。 58
(第1820
-1825
) 等蛋白质组学的可视化方法
, 国家标准修订版摩尔细胞。生物。
, 2006
,卷。 7
(第225
-230
) 等细菌RNA聚合酶结合的转录延伸因子GreB的结构和功能
, 单元格
, 2003
,卷。 114
(第335
-345
) 等MAMMOTH(从理论上获得的匹配分子模型):模型比较的自动化方法
, 蛋白质科学。
, 2002
,卷。 11
(第2606
-2621
) 等CRM1/Export1的体系结构表明了在形成核出口综合体期间如何实现合作
, 分子电池
, 2004
,卷。 16
(第761
-775
) 等利用局部归一化互相关函数快速搜索电磁密度图的三维模体
, J.结构。生物。
, 2003
,卷。 144
(第95
-103
) . 利用局部相关将畴结构对接到低温电子显微术的图谱中
, 《水晶学报》。D。
, 2000
,卷。 56
(第1332
-1340
) . 将原子模型拟合到电子显微镜图中
, 《水晶学报》。D。
, 2000
,卷。 56
(第1341
-1349
) 等蛋白质相互作用的结构观点
, 货币。操作。结构。生物。
, 2004
,卷。 14
(第313
-324
) 等从结构蛋白质组学的词汇到文献
, 自然
, 2003
,卷。 422
(第216
-225
) 等RyR1-FKBP12相互作用的结构表征
, 分子生物学杂志。
, 2006
,卷。 356
(第917
-927
) 等单个免疫球蛋白G分子在溶液中的结构和柔韧性
, 结构(拱度)
, 2004
,卷。 12
(第409
-415
) 等stutzeri假单胞菌AK61的氰化物降解腈酶是一个2倍对称的14亚单位螺旋
, 结构
, 2003
,卷。 11
(第1413
-1422
) 等通过比较建模和电子冷冻显微镜对蛋白质组分的结构表征
, J.结构。生物。
, 2005
,卷。 149
(第191
-203
) 等通过迭代比较建模和CryoEM密度拟合优化蛋白质结构
, 分子生物学杂志。
, 2006
,卷。 357
(第1655
-1668
) , . 结合电子显微镜和比较蛋白质结构建模
, 货币。操作。结构。生物。
, 2005
,卷。 15
(第578
-585
) 等评估针对CASP6比较建模类别提交的预测
, 蛋白质
, 2005
,卷。 61
补充7
(第27
-45
) 等蛋白质-蛋白质复合物低分辨率识别的系统研究
, 程序。美国国家科学院。科学。美国
, 1999
,卷。 96
(第8477
-8482
) 等SPI-EM:在3D-EM地图中预测CATH超家族的工具
, 分子生物学杂志。
, 2005
,卷。 345
(第759
-771
) 等蛋白质超家族结构变异性指导下的3D-EM柔性拟合
, 结构
, 2006
,卷。 14
(第1115
-1126
) , . 原子模型与电子显微镜重建的对接
, 方法。酶制剂。
, 2003
,卷。 374
(第204
-225
) , . 多分辨率结构的建模技巧和拟合技术
, 结构(拱度)
, 2001
,卷。 9
(第779
-788
) 等Situs:一个用于将晶体结构对接到电子显微镜低分辨率地图的软件包
, J.结构。生物。
, 1999
,卷。 125
(第185
-195
)
作者注释
©作者2006。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oxfordjournals/org






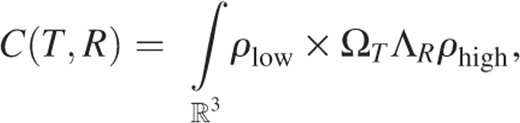
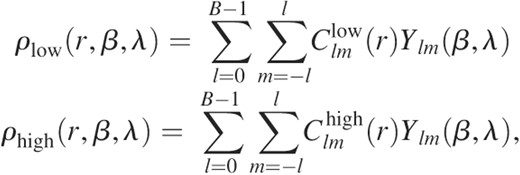
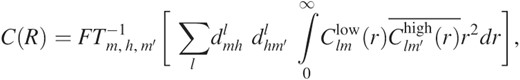


{kind=link}
{kind=link}
{kind=link}