摘要 人类基因组中的大多数CpG二核苷酸在胞嘧啶碱基处甲基化。 然而,活性基因调控元件相对于其侧翼区域通常是低甲基化的,一些转录因子(TF)的结合因其靶序列的甲基化而减少。 通过使用甲基化敏感SELEX(通过指数富集配体的系统进化)对542个人类TF进行分析,我们发现也有许多TF偏好CpG-甲基化序列。 其中大多数属于扩展同源结构域家族。 结构分析表明,甲基胞嘧啶的同源结构域特异性取决于与甲基胞嘧啶5-甲基的直接疏水相互作用。 本研究对表观遗传DNA修饰对人TF结合特异性的影响进行了系统研究,并揭示了许多发育重要的蛋白质对含mCpG序列的偏好。
胞嘧啶在CpG二核苷酸(mCpG)上的甲基化在调节人类基因组结构和活性中起着重要作用。 哺乳动物基因组中的大多数CpG二核苷酸是甲基化的,但甲基化模式并不一致。 核小体相关DNA的甲基化率低于核小体之间更容易获得的连接序列( 1 , 2 ). 此外,甲基化模式因细胞类型而异( 三 )这些变化与基因表达有关。 高表达基因的基因体高度甲基化( 4 , 5 )而活性基因调控元件的甲基化程度较低( 6 – 8 ).
DNA甲基化被认为可以直接或间接调节转录。 CpG甲基化可以通过阻止一些转录因子(TF)与其识别基序的结合来直接抑制转录[例如( 9 – 12 )]. 此外,mCpG二核苷酸可以被一类特定的蛋白质识别,即甲基CpG结构域结合蛋白,其中一些可以募集组蛋白脱乙酰酶,并被认为促进局部染色质缩合( 7 , 13 ). 人们普遍认为甲基化是重编程的障碍( 14 , 15 )TF与先前甲基化位点的结合( 16 )或去除CpG甲基化( 7 )参与细胞分化或重编程。
甲基化模式是跨细胞分裂遗传的。 这是由甲基转移酶DNMT1完成的,它与DNA复制叉结合,在模板链甲基化的位置使新合成的DNA链甲基化( 7 ). 这个过程,连同细胞质决定因子(如TF)的遗传,形成了表观遗传记忆的基础,表观遗传记忆力允许获得特征的细胞遗传,如分化状态[例如( 14 )]. TFs和DNA甲基化在表观遗传中的作用因此得到了很好的证实。 一些研究还通过分析单个TF与甲基化位点的结合来表征这些关键决定因素之间的相互作用( 9 , 10 , 12 , 17 , 18 )和/或分析多个TF与有限数量序列的结合( 19 , 20 ). 然而,迄今为止尚未对大量TF与所有可能的DNA序列的结合进行系统分析。
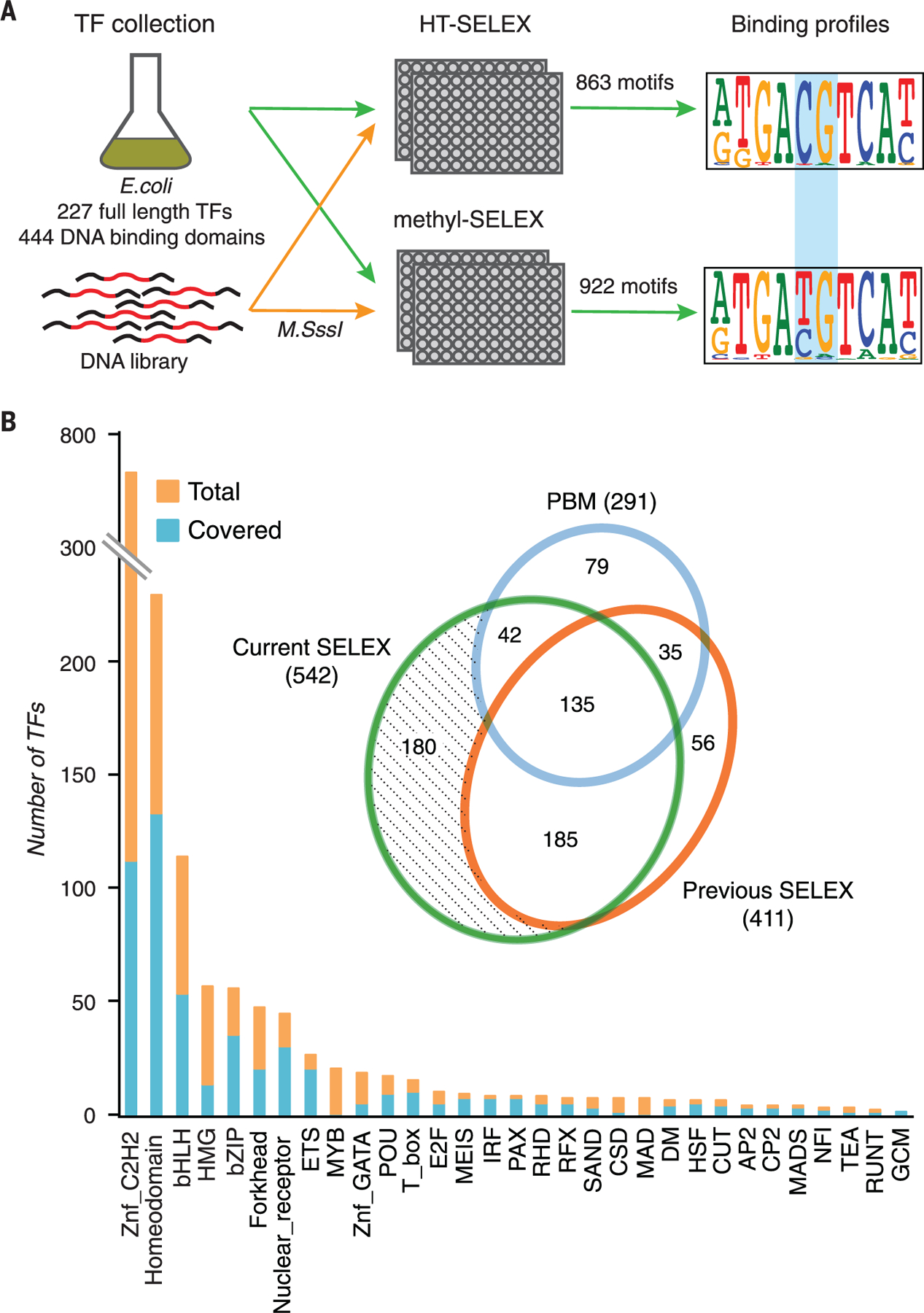
存在和不存在CpG甲基化的HT-SELEX揭示了TF结合的特异性 为了全面表征胞嘧啶甲基化对TF结合的影响,我们通过指数富集法进行了HT-SELEX[配体的高通量系统进化( 21 , 22 )]分析约1000个人类TF扩展DNA结合域(eDBDs);详细信息见 表S1 和 方法 )和约550个全长TF。 该集合包括Vaquerizas描述的84%的高置信TF 等 . ( 23 )(a类和b类; 表S1 ). 用非甲基化DNA配体和甲基化DNA配体进行检测( 24 )在每个选择周期之前使用CpG-特异性胞嘧啶5-甲基化酶M.SssI(参见 方法 详细信息; 图1A ). 配体包含40个碱基对(bp)的随机序列,并在分析之前和每个选择周期之后进行测序。 使用前面描述的Autoseed管道对所得数据进行分析,Autoseed是一种基于不同种子序列识别的从头绑定基序发现方法,这些种子序列随后用于生成位置权重矩阵(PWM)模型[参见( 25 )和 方法 ]. 我们( 21 , 26 , 27 )和其他人之前已经确定,使用HT-SELEX生成的基序与使用其他最先进方法获得的基序类似,例如蛋白质结合微阵列或细菌单杂交分析( 28 , 29 )和根据其预测体内TF结合的能力而具有生物学相关性( 30 – 32 ).
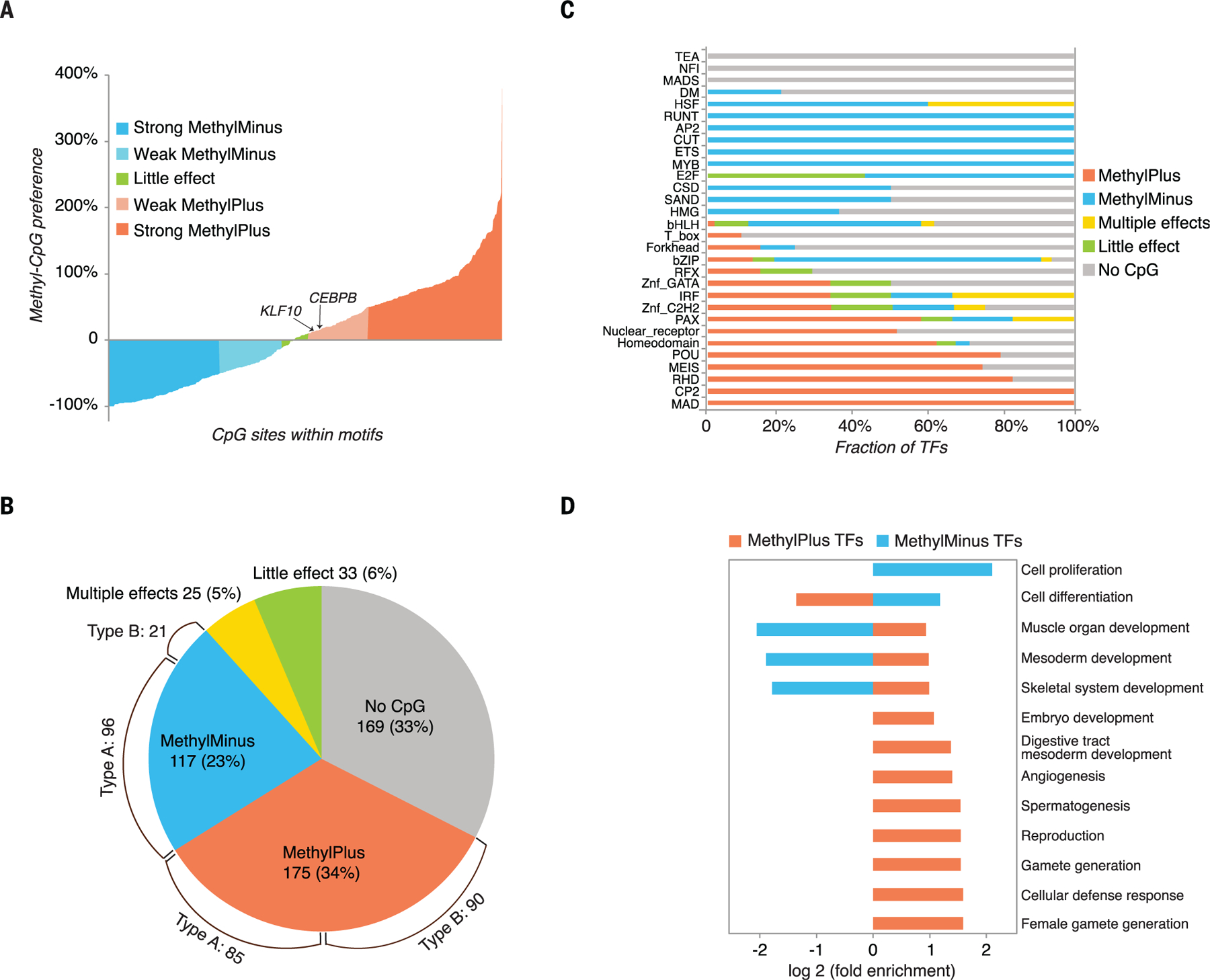
图1。 甲基-SELEX。
(A) SELEX过程的示意图,该过程允许识别所有DNA序列的转录因子结合特异性,包括含有甲基化和非甲基化CpG二核苷酸的序列。 该过程使用两个平行反应,一个是非甲基化DNA(顶部,HT-SELEX),另一个是在每个选择周期甲基化的DNA(底部,methyl-SELEX)。 显示了获得基序的全长TF和扩展DBD的数量。 蓝色矩形表示受甲基化影响的CpG二核苷酸的位置。 (B) 家庭TF覆盖率。 插图是一张维恩图,比较了本研究与之前使用蛋白结合微阵列(PBM)进行的大型研究中哺乳动物TF的覆盖率( 35 , 36 )和HT-SELEX( 21 , 25 , 26 ). Znf,锌指。
与其他方法相比,HT-SELEX能够检测更长的结合基序,因为输入库的复杂性很高( 26 ). 虽然SELEX测量的是序列的富集度,而不是结合本身的亲和力,但序列富集的顺序与其亲和力的顺序相同。 此外,我们之前已经表明,从早期HT-SELEX循环中获得的基序与通过更直接测量亲和力的方法获得的基模类似,例如寡核苷酸竞争分析( 33 )或比较单个SELEX循环中富集度的测定( 26 ). 因此,这里提出的基序可以直接用于使用阈值的基序匹配,其中只有亲合等级影响结果。 然而,获得的分数应被视为估计值,而不是真正的亲和力度量值。
每个TF的模体发现中位数成功率为47%,总共获得了444个eDBD和227个全长TF的数据( 表S2 ). 如前所述( 26 )C2H2锌指蛋白、SMAD蛋白和SANT/Myb蛋白的成功率相对较低,这可能是因为一些C2H2蛋白的识别基序很长,SMAD蛋白充当专性异源三聚体, 以及许多SANT/Myb蛋白被误分类为TF,尽管它们缺乏DNA结合所需的关键氨基酸( 25 , 34 ).
TF家庭的平均覆盖率为60%( 图1B ). 单个TF的覆盖率远高于以往系统研究中报告的覆盖率( 26 , 35 – 37 ). 例如,本研究使用非甲基化和甲基化配体,以及我们之前发布的HT-SELEX数据( 26 )分别恢复了542和411个TF的模型,代表343和239个明显不同的特异性( 图2 和 表S2 ). 本研究中获得的基序与先前研究的TFs的早期数据高度一致( 图S1 和 S2A公司 ). 与之前确定的任何HT-SELEX基序不同的大多数基序是C2H2锌指蛋白的基序,其特异性尚未确定,或代表先前未识别出对mCpG的偏好的已知TF,例如本研究中为同源域蛋白集合新确定的基序( 图S2 ). 总的来说,HT-SELEX型号目前适用于约1400台中的632台( 23 )人类TF。
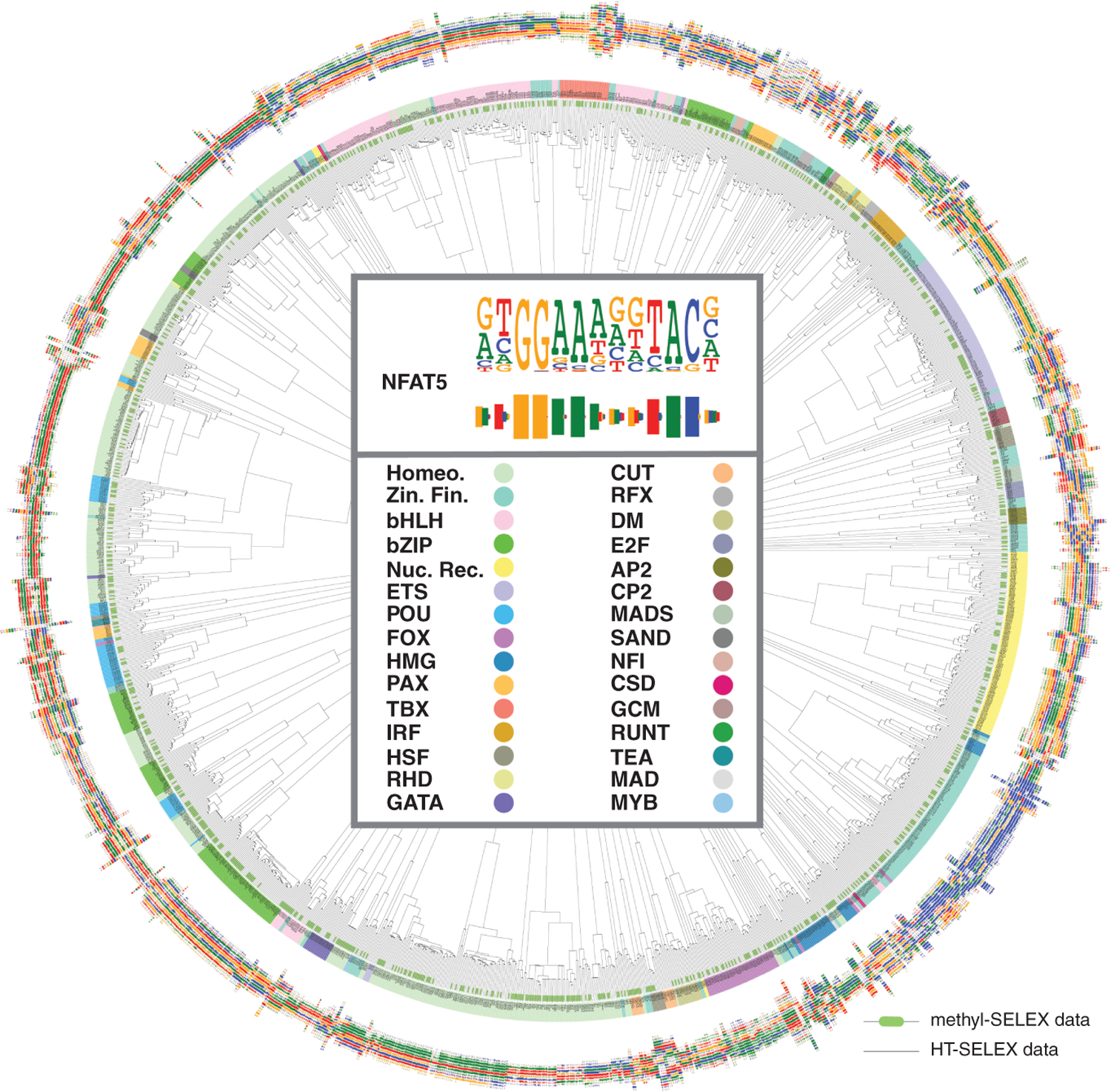
图2。 图案的相似性。
树状图表明HT-SELEX(细树状图线)和methyl-SELEX(树枝状图线末端的粗绿色条)的基序之间的相似性。 条形码徽标( 25 )对于每个因素,也显示了。 树状图的中心显示了将序列徽标转换为条形码徽标(顶部)和TF系列的颜色键(底部)的示例。 同一结构家族中TF的基序通常彼此相似,而甲基-SELEX和HT-SELEX-的基序在大多数情况下也密切相关(绿色和黑色末端出现在同一分支中)。 这是因为许多TF的基序中没有CpG,甲基化引起的变化通常只影响基序中的一个二核苷酸。 荷马。, 同源结构域; 锌。 翅片。, 锌指; 核子。 rec.,核受体。
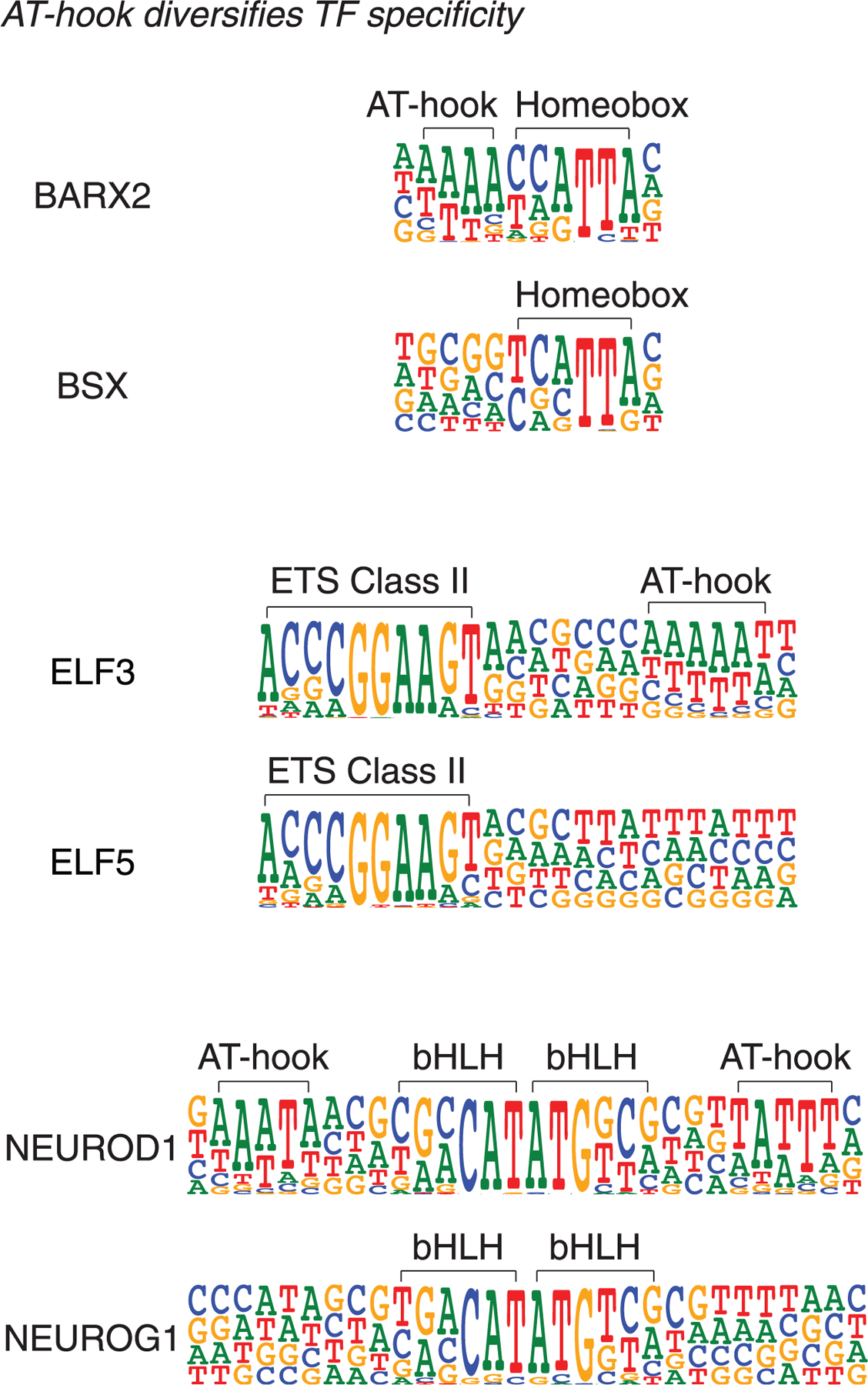
对新的HT-SELEX数据的分析还揭示了TF结合特异性的进化机制,BARX同源结构域、ELF3 ETS家族TF和bHLH家族TF NEUROD1利用了该机制,其中在主DBD旁边添加一个AT-hook结构域会优先选择侧翼的AT-rich序列( 图3 ).
图3。 通过AT-hook添加使Paralog的特异性多样化。
通过添加AT-hook肽基序说明了TF结合特异性的演变。 同源域TF BARX2、ETS因子ELF3和bHLH蛋白NEUROD1的特异性与相关TF不同,因为添加了识别短的富含AT的氨基酸序列的类AT-hook氨基酸序列。
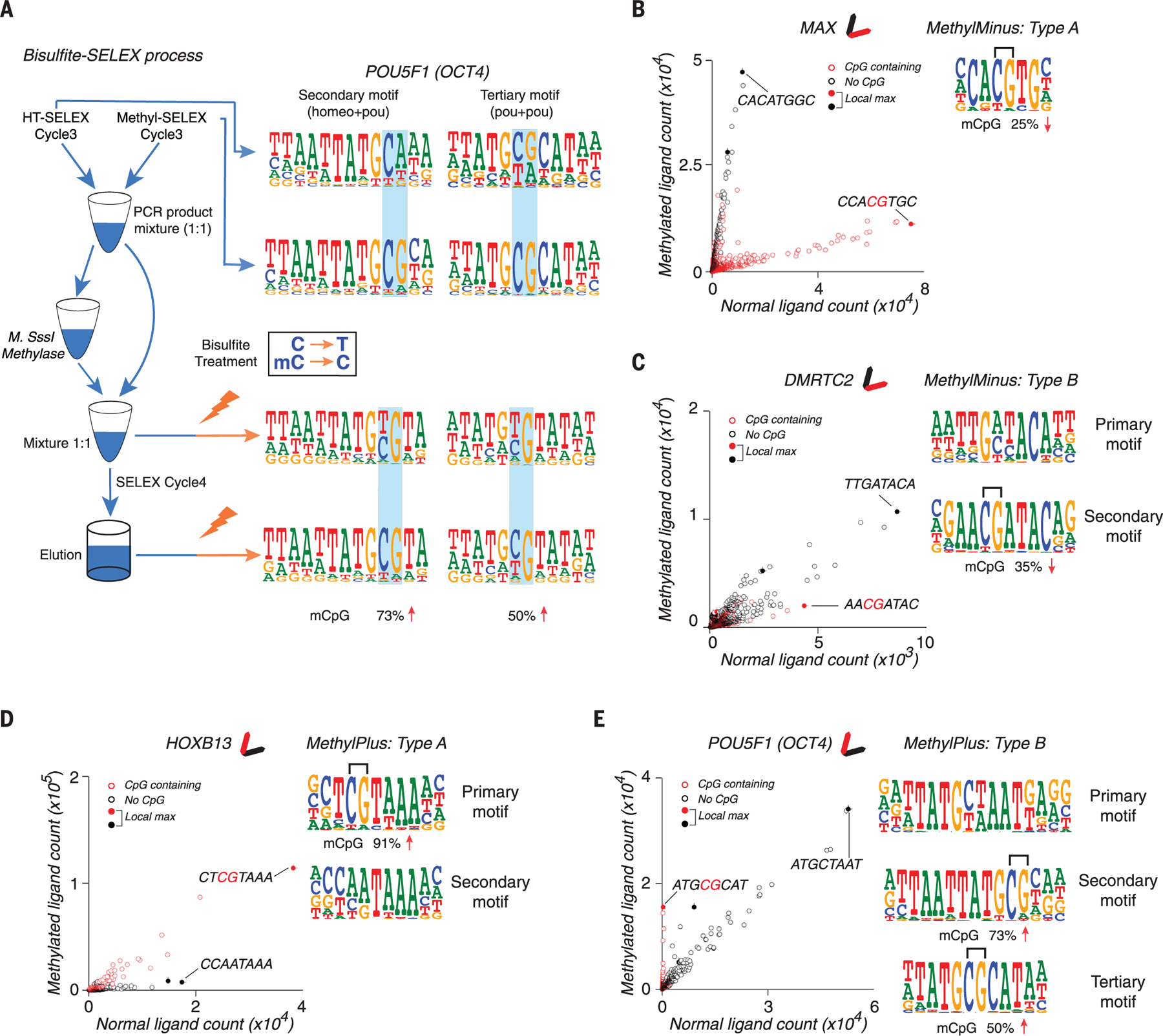
CpG甲基化对TF-DNA结合有重要影响 使用甲基-SELEX工艺(请参阅 方法 ),如果TF富集了含有和不含CpG二核苷酸的两个序列,则可以确定CpG甲基化对TF结合的影响。 然而,很难确定甲基化对TF的影响,TF对其基序中的CpG有很强或绝对的需求,因为它们可以产生相同的含CpG基序,或者在DNA甲基化时无法产生任何基序。 为了测量这种情况下CpG甲基化的影响并验证甲基-SELEX的结果,我们将识别基序包含CpG序列的大多数TF置于一个亚硫酸氢盐-SELEX-循环中( 图4A ; 请参阅 方法 ). 使用这种方法,可以在一个反应中确定TF对其含有CpG的识别序列的非甲基化或甲基化形式的偏好。 亚硫酸氢盐SELEX的结果证实了甲基-SELEX的大多数结果,并且由于其更高的灵敏度,亚硫酸氢酯SELEX还揭示了许多其他TF对非甲基化或甲基化CpG的偏好(有关比较,请参阅 表S3 ).
图4。 mCpG对TF结合的影响示例。
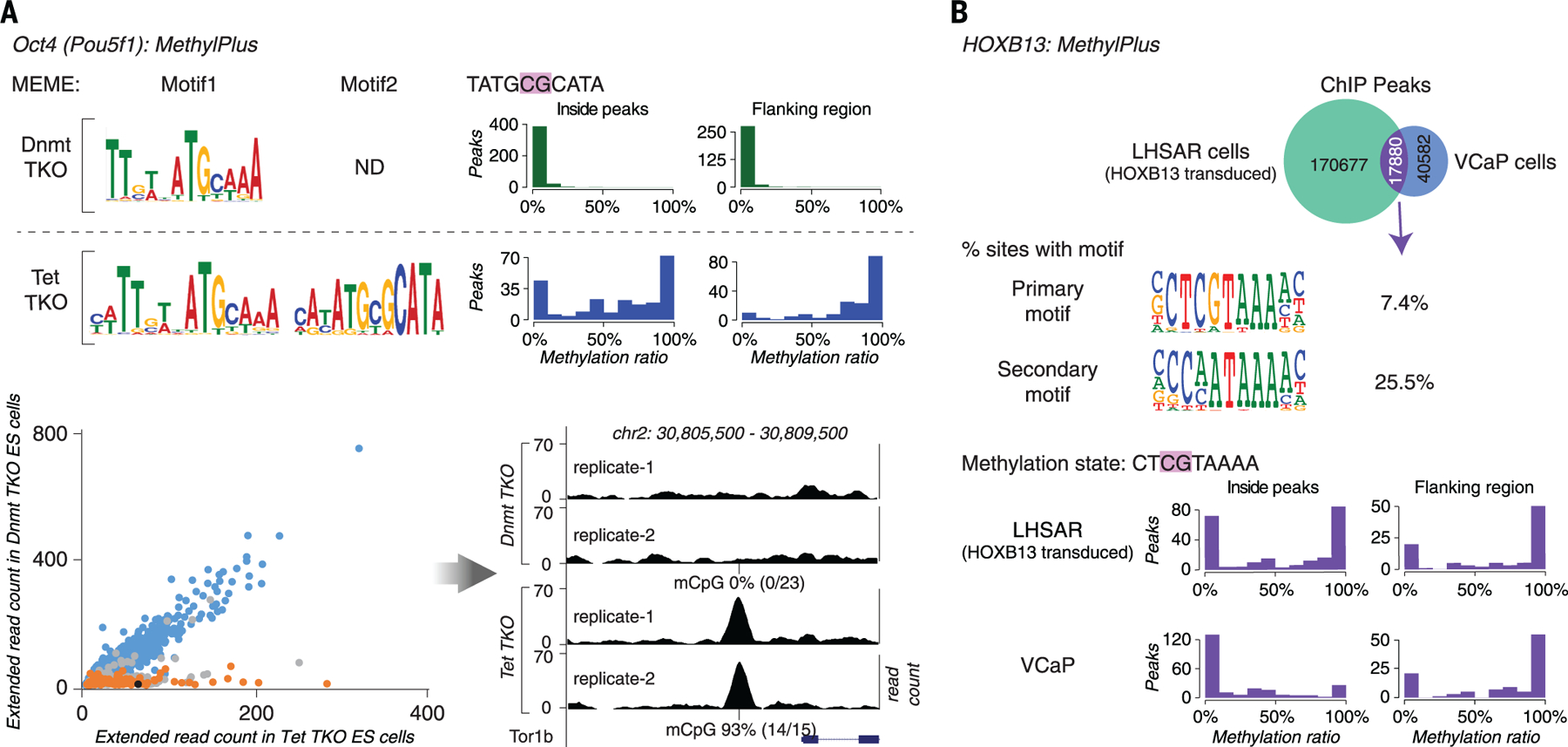
(A) 亚硫酸氢盐SELEX。 从亚硫酸氢盐-SELEX工艺的不同阶段回收了POU5F1(OCT4)的两种模型。 OCT4可以与对应于所示基序的非甲基化和甲基化序列结合,但它更喜欢在所示CpG甲基化时结合序列(如方框所示,亚硫酸氢盐处理后仍为CpG)。 闪电表示亚硫酸氢盐处理,蓝色阴影突出显示受甲基化影响的二核苷酸。 底部的数字显示了从周期3到周期4 mCpG的增加百分比。 ( B类 )a型甲基分钟TF,MAX(Myc-associated factor X)的示例。 散点图(左)显示来自甲基SELEX的所有8聚体子序列的计数( 年 轴)和HT-SELEX( x个 轴)。 实心圆圈表示在Huddinge距离内比任何其他子序列更丰富的子序列( 25 )也指出了富集程度最高的序列(CCACGTGC)。 由于CpG的甲基化抑制MAX结合,红圈群体(具有CpG的序列)形成位于黑圈群体(没有CpG的序列)下方的细长图案; 这也通过简化的字形(顶部)显示。 当与最佳位点的结合被阻断时,其他结合较弱的序列(CACATGGC)富集得更强烈。 MAX图案的标志也如图所示(右图),其下方显示了亚硫酸氢盐-SELEX中CpG甲基化的影响。MAX被归类为A型,因为其图案的一致性包含CpG(括号)。 ( C类 )一种B型甲基-分钟TF,DMRTC2,其主基序(右上)不受甲基化的影响,但次基序(左下)中的CpG受甲基化影响。与两个基序一致的序列显示在散点图上。 ( D类 和 E类 )如(B)和(C)所示,但对于A型甲基-plus TF HOXB13(D)和B型甲基-pus TF POU5F1(OCT4)(E)。 在CpG甲基化存在的情况下,POU5F1(OCT4)使亚序列ATGCGCAT更加丰富。 OCT4还丰富了不含CpG且不受甲基化影响的子序列ATGCTAAT。
结合甲基SELEX和亚硫酸氢盐SELEX的结果表明,一些TF不识别具有CpG序列的位点,因此它们的结合不受CpG甲基化的影响( 图S3A ; “无CpG”类)。 第二类TF识别含CpG的序列,但CpG甲基化对结合几乎没有影响( 图S3B ; “小效果”类)( 38 , 39 ). 第三类TF与它们的识别序列的甲基化版本没有结合,或结合更弱( 图4 , B类 和 C类 、和 图S3C ; “甲基-分钟”类)。 在大多数情况下(82%),甲基化影响了富集度最高的基序(初级基序)得分最高的序列(一致序列)。 这些TF被归类为甲型( 图4B 和 图S4 ; 请参阅 方法 详细信息)。 在其余情况下,主要基序的一致序列不包含CpG,但TF与其他含有CpG的富集位点的结合可能会受到甲基化的影响。 这些TF被归类为B型甲基三聚氰胺( 图4C 和 图S5 ). 甲基缺失基团包括几种蛋白质,其先前存在抑制mCpG结合的证据( 9 , 10 , 12 , 17 , 18 ) ( 图S6A )表明甲基-SELEX法具有较高的敏感性和特异性。
除了先前已知的CpG甲基化对TF结合的中性和负面影响外,我们还发现了第四类TF,它们倾向于在相应的非甲基化序列上与某些甲基化序列结合( 图4 , D类 和 E类 、和 图S3D ; “甲基-plus”类)。 这一类包括以前报告过的弱偏好mCpG的TF,如CEBPB( 40 ),荷兰皇家空军( 19 , 20 , 41 )和RFX5( 20 ) ( 图S6 , B类 到 D类 ). 此外,我们鉴定了许多TF,在我们的分析中,它们比这些蛋白质对mCpG表现出更强的偏好性( 图S6D 和 表S3 ). 在这类TF中约有一半的TF中,甲基化影响了主要基序的一致序列( 图4D 和 图S7 ; 甲基+A型; 请参阅 方法 详细信息),在其余情况下,具有mCpG的较弱位点优先结合于相应的未甲基化位点( 图4E 和 图S8 ; 甲基+B型)。 为了通过不同的实验方法验证结果,我们进行了酶促甲基化( 11 )蛋白质结合微阵列( 35 ); 该分析证实了基于甲基-SELEX的八项任务中的七项( 图S9 ).
为了确定单个CpG甲基化对TF结合的影响,我们计算了亚硫酸氢盐-SELEX实验期间基序中特定位置mCpG增加的百分比(参见 方法 ). 单个CpG位点的甲基化通常影响TF结合,影响范围为−100到+380%( 图5A ). 观察到高亲和力位点和中等亲和力位点的影响( 图S4 , 第5章 , 第7部分 、和 第8节 和 表S3 ). 因此,取决于是否存在CpG二核苷酸,许多TF可以在生物相关亲和力范围内结合到甲基化敏感和不敏感位点。
图5。 基于甲基-SELEX和亚硫酸氢盐-SELEX.的TF分类。
(A) 单个CpG二核苷酸甲基化对人TF结合的影响。 显示了在一轮亚硫酸氢盐-SELEX期间TF结合基序中所有mCpG二核苷酸的百分比增加。 大多数CpG的甲基化对TF结合有负面(蓝色)或正面(橙色)影响。 ( B类 )基于甲基SELEX和亚硫酸氢盐SELEX数据联合分析的TF分类(参见 表S3 了解每个因素的详细信息)。 饼图显示了不受胞嘧啶甲基化影响(无CpG或几乎没有影响)、偏好非甲基化CpG(甲基-微量)或偏好甲基化Cp G(甲基-plus)的TF的比例。 此外,25个TF在其结合序列的不同位置或不同基序(多重效应)上对mCpG二核苷酸表现出不同的偏好。 可以与多个基序结合的转录因子根据含有CpG二核苷酸的基序进行分类,如果存在这样的基序的话。 括号表示甲基-分钟和甲基-plus基团的A型和B型TF的数量。 ( C类 )每个结构TF家族中各组TF的分数。 ( D类 )甲基-plus和甲基-minus TFs的基因本体富集分析。 显著(修正)的生物工艺等级 P(P) <0.005)富集或耗尽(基于获得基序的所有TF,相对于随机期望,超过两倍)。
CpG甲基化会影响大多数TF的结合 接下来,我们通过结合亚硫酸氢盐-SELEX和甲基-SELEX数据对所有TF进行分类。 在TF与两个或更多基序结合的情况下,分类基于包含CpG二核苷酸的基序(参见 方法 , 表S3 、和 数据S1 和 S2系列 了解每个TF的详细信息)。 该分析表明,在519个可分类的TF中,60%可以与一个或多个高度或中度(最大值>10%)富集的序列结合,这些序列的富集受CpG甲基化的影响( 图5B ); 在这些TF中,117个TF的结合被抑制(23%),175个TFs的结合被增强(34%),25个TF(5%)对不同的基序或单个基序中不同的CpG位置表现出不同的效应(“多重效应”类别; 方法 和 表S3 ). 其余40%的转录因子不受CpG甲基化的影响( 图5B ). 其中,169个TF(33%)在其识别序列中没有CpG二核苷酸,33个TFs(6%)可以与含有CpG的基序结合,但对甲基化或非甲基化CpG没有明显的偏好。 在本分析中,根据之前报告CEBPB偏好的数据,使用了±10%的影响阈值( 40 )和多种KLF蛋白( 41 )对于mCpG。 TFs分为五类(甲基-微量、甲基-多效、小效和无CpG)是稳健的; 在129例获得了相同TF全长和eDBD结构数据的病例中,125例被分类为类似的( 数据S2 ). 使用独立表达式构造的重复实验也证实了分析的稳健性( 图S10A ).
TF家族在CpG甲基化敏感性上存在差异 接下来,我们比较获得的基序,以确定特定TF结构家族是否具有共同特征。 该分析表明,bHLH-、bZIP-和ETS-家族TF通常被mCpG抑制,而NFAT(RHD)因子和扩展同源域家族的许多成员(例如同源域、POU和NKX)倾向于结合到含有mCpG-的序列( 图5C 和 数据S2 ). 然而,家族内存在差异,一些bZIP蛋白与含有mCpG的位点结合,亲和力不变或稍高( 图5C 和 数据S2 ). 类似地,除了典型的TAATTA共有基序外,许多(但不是所有)同源域蛋白还与甲基化序列TmCGTTA结合( 数据S2 和 表S2 ). 一般来说,甲基+TF通常倾向于结合到彼此不同的非甲基化和甲基化位点( 图S10B 和 数据S2 ). 在几乎所有情况下,观察到的差异都是由于一个或多个CpG二核苷酸频率的变化所致( 数据S2 ). 相反,其他基团的HT-SELEX和甲基SELEX位点彼此非常相似( 图S10B 和 数据S2 ).
为了便于对不同的结合特异性进行全局分析,我们生成了一个代表性图案的最小集合,在给定相似性边界的情况下,可以代表整个集合中的所有图案[参见( 26 , 42 )和 方法 有关详细信息]。 该分析表明,CpG甲基化可以抑制与42个代表性结合基序的结合( 图S11A ); 在所有这些中,受影响的CpG被纳入共识序列。 此外,当CpG甲基化时,总共有44个代表性的结合基序优先结合( 图S11B ); 其中,除四个外,其余均在基序一致性中具有受影响的CpG。 对哺乳动物基序匹配的保守性分析表明,这两组新发现的许多基序都是保守性的,这表明它们在生物学上是相关的( 图S11 和 第12节 ). 考虑到CpG序列上的高突变负载,观察到的守恒性尤其引人注目( 图S12C )[例如( 43 )].
对mCpG抑制结合的TF进行Panther GO-slim基因本体富集分析,发现细胞增殖和细胞分化是最富集的生物过程( 图5D ). 相反,偏好mCpG的TF通常参与胚胎和器官发育过程( 图5D ). 这种富集分析可以确定特定类别的TF相对于我们获得基序的所有TF是否富集。 然而,TF富集的原因无法通过这种方式确定。 富集可能是由于许多同源TF可能从一个原始TF遗传了其生物作用和甲基化特异性。或者,富集可能是生物驱动的,例如, 以确保参与细胞增殖的TF不能结合和激活位于控制发育的基因上的甲基化和沉默调节元件,相反,参与胚胎发育的TF能够结合甲基化位点并诱导细胞染色质状态的重大变化。 在这方面,需要注意的是,大多数甲基+TF属于发育TF的一个关键家族,即同源结构域蛋白。 示例包括指定胚胎前后轴的同源结构域因子(例如HOXC11和HOXB13)、定义发育过程中细胞谱系的NKX蛋白以及多能性调节器POU5F1(OCT4)( 44 – 48 ).
CpG甲基化对体内TF结合的影响 为了确定TF在体内是否也显示出预期的偏好,我们使用现有的染色质免疫沉淀测序(ChIP-seq)数据和新的ChIP-exo酶(ChIP-exo)实验来定位关键TF,并使用全基因组亚硫酸氢盐测序来鉴定两种人类结直肠癌细胞系LoVo和GP5d中的mCpG序列。 结果与体外分析基本一致( 数据S3 和 表S4 ). 然而,在每种情况下,TF占据位点的甲基化水平都低于其相应的侧翼区域。 此外,一些甲基+TF结合的位点缺乏甲基化。 这些结果与早期的发现一致,即TF结合可能通过TET/TDG依赖的去甲基化途径导致局部DNA甲基化的丢失( 7 , 49 , 50 ).
由于TF结合诱导的甲基化状态变化可能会混淆ChIP-seq分析,因此我们接下来在甲基化受到干扰的三种不同条件下在体内测试TF结合。 首先,我们将HOXC11位点引入荧光素酶报告子结构,否则该结构完全不含CpG二核苷酸。 HOXC11与非甲基化和甲基化报告子结构的共转染表明,正如甲基-SELEX预期的那样,识别序列的甲基化导致转录活性增加( 图S13A ). 在第二组实验中,我们分析了小鼠胚胎干细胞(ES)体内显示CpG甲基化水平升高或降低的TF结合。 为了产生具有高CpG甲基化水平的ES细胞系,我们使用CRISPR-Cas9删除所有三种TET酶。 全基因组亚硫酸氢盐测序显示该细胞系中脱氧核糖核酸酶(DNase)I超敏位点甲基化水平增加( 图S13C )与对照细胞或之前描述的缺乏CpG甲基化的ES细胞系相比[ Dnmt公司 三次击倒( Dnmt公司 -TKO)细胞系( 51 )]. 然后,我们对所有三个ES细胞系中的甲基+TF OCT4和甲基-分钟TF n-Myc进行了ChIP-seq分析。 该分析表明甲基化状态的改变导致了TF结合的预期变化( 图6A ; 图S13 , B类 到 D类 、和 S14标准 ; 和 数据S3 , C类 和 D类 ). 除了甲基化增加外 泰特 -TKO细胞系也缺乏5-羟甲基胞嘧啶,这已被证明能特异性影响TF结合( 52 ). 然而,在这项研究中观察到的影响不太可能是由于这种修饰碱基的缺失,因为它以较低的等位基因频率存在( 53 )和OCT4与含CpG基序的基序结合的差异也在两组的比较中观察到 Dnmt公司 -TKO和野生型细胞系( 图S14C ).
图6。 ChIP-seq分析。
(A) OCT4在体内偏好甲基化基序。 对缺乏甲基胞嘧啶的ES细胞进行OCT4的ChIP-seq分析( Dnmt公司 -TKO)或显示基因调控区甲基化增加( 泰特 -TKO)。 MEME的基序富集分析(左上)仅从 泰特 -TKO细胞。 含有基序2的大多数OCT4占据位点在 泰特 -TKO细胞(蓝色直方图),但不在 Dnmt公司 -TKO细胞(绿色直方图)(右上角)。 散点图(左下)显示了基序匹配位置的ChIP扩展读取覆盖范围 泰特 - ( x个 轴)和 Dnmt公司 -TKO公司( 年 轴)单元格。 基序1匹配位置(蓝色)的ChIP-seq峰高在细胞类型中相似,而基序2匹配位置的峰高在甲基化状态改变(橙色)的细胞中较高 泰特 -TKO细胞。 只分析与两个或多个亚硫酸氢顺序读取重叠的站点。 含有基序2的峰与甲基化没有变化或变化小于截止值(从≤20% Dnmt公司 -TKO≥80%英寸 泰特 -TKO)为灰色。 黑点表示右下面板中显示的示例峰值位置。 ( B类 )外源性引入的HOXB13与原代前列腺上皮细胞系LHSAR中的甲基化位点结合。 对表达HOXB13的慢病毒转导的VCaP前列腺癌细胞和LHSAR细胞进行HOXB12的ChIP-seq分析。 对两种细胞株共有的峰的分析(顶部)表明,HOXB13可以结合到两个不同的基序,其中一个基序(SELEX初级基序)通常包含CpG二核苷酸。 大多数含有CpG的共有峰的位置在LHSAR细胞中被甲基化,表明HOXB13可以与甲基化位点结合。 占据位点的甲基化水平通常很低或很高,这与特定等位基因存在或不存在甲基化的事实一致。 VCaP前列腺癌细胞中的甲基化较低,可能是因为结合诱导的去甲基化( 7 ).
最后,为了排除组成结合TF甲基化状态改变的影响,我们分析了甲基+TF HOXB13的体内结合特异性,该甲基+TF-HOXB13是外源性导入不表达该蛋白的前列腺上皮细胞系。 以前的研究表明,在外源性HOXB13和FOXA1表达后,正常前列腺上皮细胞的染色质状态变得与前列腺癌细胞的染色素状态更为相似( 54 ). 与甲基SELEX的证据一致,来自HOXB13转导的细胞的ChIP-seq分析显示,HOXB13在体内与含有mCpG的位点有很强的结合( 图6B , 图S15 、和 数据S3C ). 在48小时的潜伏期内,结合位点的甲基化状态没有受到强烈影响( 图S15 ). 然而,在前列腺癌细胞系VCaP中,相应的峰值显示甲基化水平较低,这表明这些区域甲基化水平降低可能有助于肿瘤发生过程中染色质的重新编程( 图6B , 图S15 、和 数据S3C ). 与HOXB13也与非甲基化CpG结合的能力一致,尽管亲和力较低,但它似乎能够在体内与许多非甲基化位点保持结合。 mCpG位点在未甲基化之前是否存在短暂结合的子集,尚待确定。 然而,人们很容易推测,这些位点可能在过渡细胞状态中具有特定的生物学作用,例如在短期发育的祖细胞或在成体传递放大细胞群中。 三种类型的TF甲基化偏好(正、负和小影响)、两种甲基化状态(mC和C)以及TF结合甲基化状态的三种可能后果(甲基化、去甲基化或无变化)表明其他机制( 表S5 )因为表观遗传状态的增强以及负反馈和正反馈可能有助于生物过程。
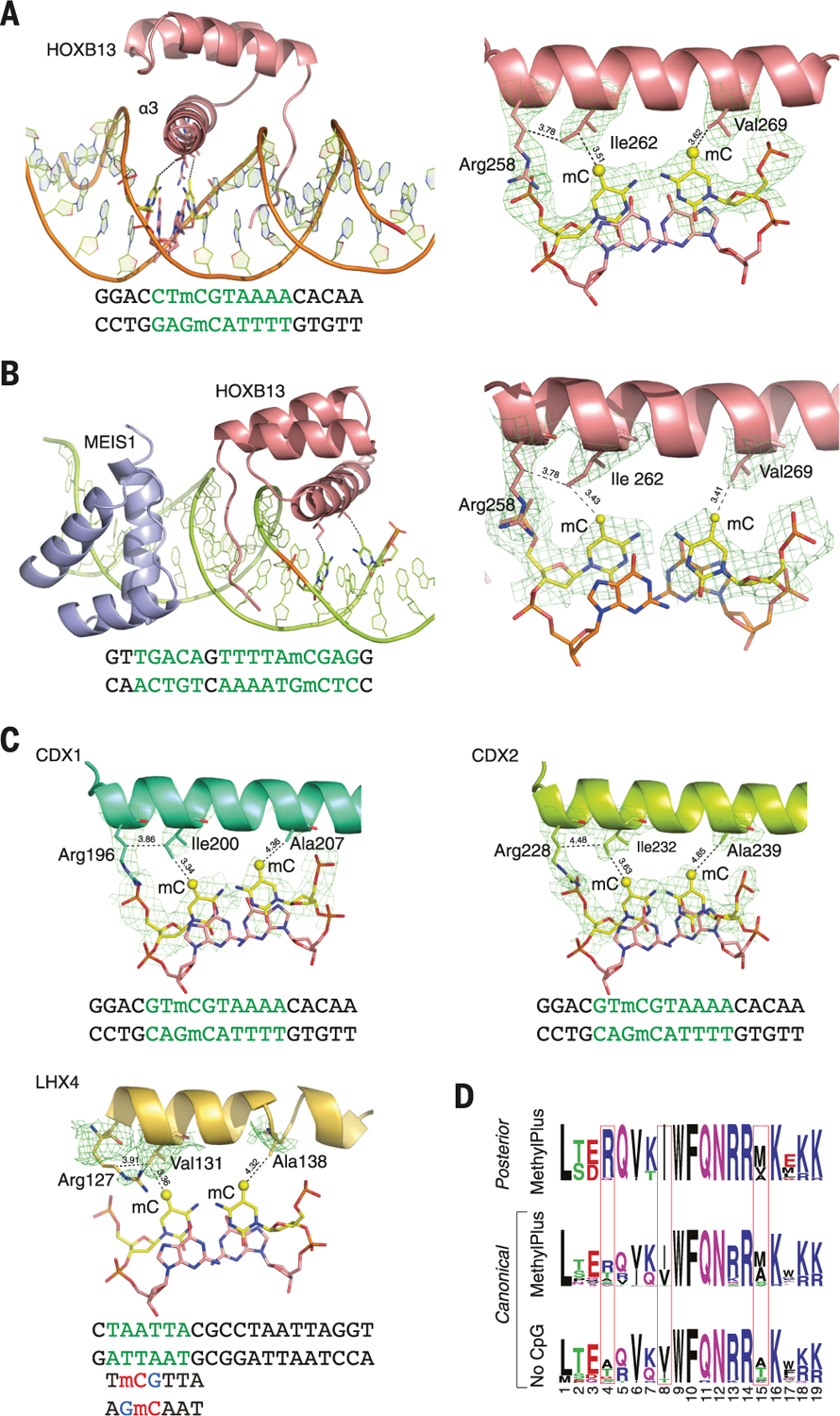
mCpG偏好的结构基础 为了验证我们的发现并确定观察到的同源域TF对甲基化胞嘧啶偏好的分子基础,我们在存在或不存在异二聚体伴侣MEIS1的情况下,解决了HOXB13与其首选位点CTCGTAAA的双重甲基化版本结合的结构。 这些蛋白表达于 大肠杆菌 纯化并结晶结合到含有单体CTmCGTAAA和异二聚体CTmCCTAAAAcTGTCA基序的合成双链DNA片段。 求解结构并将其与之前求解的HOX蛋白质结构进行比较( 55 – 60 )揭示了一个非常相似的DBD架构,其预期核心由三个α螺旋组成( 图7A 和 图S16 ). 与所有已知的同源结构域结构一样,HOXB13 DBD的两个部分与DNA相互作用:紧密包裹在主沟中的识别螺旋α3,以及与次沟相互作用的N末端尾部( 图7A ; 3.0-Ω分辨率)。 对DNA接触的分析表明,HOXB13通过CpG二核苷酸的两个甲基胞嘧啶的氨基酸和5-甲基之间的直接疏水相互作用来识别mCpG。 HOXB13伊利 262 与第一甲基胞嘧啶形成疏水性接触,而缬氨酸 269 识别与TCG序列鸟嘌呤相反的第二甲基胞嘧啶( 图7A ). 此外,Arg的脂肪族链 258 与Ile互动 262 并促成了该区域的疏水环境。 疏水相互作用也存在于HOXB13:MEIS1-DNA结构中,这表明这两种胞嘧啶的甲基在多种生理相关环境中被HOXB13强烈识别( 图7B ; 2.54-Ω分辨率)。
图7。 同源域蛋白识别mCpG的分子基础。
(A) HOXB13与甲基化DNA结合的结构揭示了后同源结构域蛋白识别甲基化胞嘧啶的机制。 左边显示的是与甲基化DNA结合的HOXB13的整体结构。 识别甲基化CpG的残基显示为球棒模型,结晶中使用的DNA序列显示在结构下方。 右侧显示的是HOXB13 DBD识别螺旋残基的复合省略电子密度图,该残基与两种甲基化胞嘧啶形成疏水相互作用。 模型的触点用虚线表示; 数字是以埃为单位的距离。 伊勒 262 与TmCG序列的mC相互作用,而Val 269 与来自互补链的mC相互作用。 精氨酸的脂肪族链 258 也有助于局部疏水环境。 绿色字母突出显示了TF特别约束的基础。 ( B类 )HOXB13概述:MEIS1异二聚体与甲基化DNA结合。 HOXB13为粉红色,MEIS1为蓝色,甲基化碱基对显示为球棒模型,触点显示为虚线,残留物和甲基化碱被标记。 与HOXB13单体类似,这两种甲基化胞嘧啶分别被Ile识别 262 和Val 269 . ( C类 )复合物省略电子密度图显示识别mCpG的CDX1、CDX2和LHX4残基。 ( D类 )序列标志显示强甲基加后同源结构域蛋白和偏好或不结合mCpG的经典同源结构域之间的相似性。 结构分析(盒子)确定的位置的残基的相同性解释了这些蛋白质相对于mCpG的不同偏好。 氨基酸残基的单字母缩写如下:A,Ala; C、 半胱氨酸; D、 天冬氨酸; E、 谷氨酸; F、 苯丙氨酸; G、 甘氨酸; H、 他的; 一、 伊利; K、 赖氨酸; L、 亮氨酸; M、 已见面; N、 Asn; P、 专业; Q、 甘氨酸; R、 精氨酸; S、 序列号; T、 Thr; 五、 缬氨酸; W、 Trp; 和Y,Tyr。
为了确定HOXB13与非甲基化和甲基化位点结合的热力学参数,我们进行了等温滴定量热法(ITC)实验。 HOXB13与非甲基化位点结合,自由能变化类似(ΔG=−10015 cal/mol; 图S17A )之前关于这类蛋白质的报道( 61 ). 与亚硫酸氢盐SELEX和甲基SELEX数据一致,与甲基化位点的结合更强(ΔG=−10824 cal/mol)。
为了确定同源结构域对mCpG的识别机制是否通用,我们解决了另外三种同源结构域蛋白质的结构:CDX1、CDX2和LHX4( 图7C ; 分别为3.2-、2.7-和2.7-Ω决议)。 CDX1和CDX2结合到其首选的GTmCGTAAA位点的结构表明,它们还通过使用相同相对位置的氨基酸直接识别甲基胞嘧啶的5-甲基( 图7C ). 虽然后型副HOX蛋白CDX1和CDX2在其TmCGTAAA基序中与mCpG强烈结合,但LHX4与典型的TAATTA位点结合,并且与含有mCpG-的序列TmCGTTA的结合较弱,在甲基SELEX或ITC实验中未检测到与非甲基化TCGTTA位点的结合( 图S17C 和 数据S2 ). 结合到典型TAATTA位点的LHX4的结构显示疏水残基Val 131 和Ala 138 在TCGTTA序列的两条链中适合与甲基胞嘧啶形成疏水接触的位置。 精氨酸的脂肪族链 127 也支持疏水相互作用( 图7C ). 这三个残基在所有LHX蛋白中都是保守的,这解释了它们对mC的强烈偏好。相反,在DLX3中,一个对mC表现出较弱偏好的同源域,对应于Arg的关键残基 127 和Ala 138 分别被苏氨酸和丝氨酸取代,导致疏水性下降( 图S18A 和 数据S2 ). 此外,TLX2完全不与TmCGTTA结合,整个结合位点的疏水性完全丧失( 图S18B 和 数据S2 ).
对不同同源结构域的氨基酸序列进行分析,这些同源结构域与含有mCpG的位点结合或不结合,证实了位于识别螺旋起点和终点的三个残基在mCpG-识别中的关键作用( 图7D ). 对甲基+TFs的结构分析,包括NKX-、IRX-和NFAT-家族蛋白,进一步证实了甲基胞嘧啶的偏好是基于其5-甲基的疏水相互作用( 图S18C ). 由于胸腺嘧啶和甲基胞嘧啶的形状相似,大多数优选mCpG的TF也结合到含有二核苷酸TG或AA的位点。 结构分析也证实了胞嘧啶甲基化抑制TF-DNA结合的机制; 在所有检查的案例中,甲基化的负面影响都是由于空间位阻( 图S18D ).
结论 利用模型生物、细胞系和体外重建系统进行的生物化学研究和实验发现了控制基因表达的主要机制。 因此,基因组在基因表达的时间和地点所传递的语言在概念层面上是可以理解的。 然而,阅读基因组说明书还需要了解所有TF的结合特异性,这些TF构成了基因调控语言的词汇。 在这项工作中,我们使用非甲基化和CpG-甲基化DNA配体对全长TF和eDBDs的DNA结合特异性进行了系统分析。 与我们之前的工作相比( 26 )这项分析使用了一个扩展的克隆集合,并对222个没有HT-SELEX基序的TF的结合基序进行了鉴定。 此外,一种改进的计算流水线( 25 )使我们能够识别57个TF的二级绑定配置文件。 综合起来,完整的数据集包含596个先前未知的基序( 表S2 )大大扩展了TF绑定特性的已知词典。
该结果还揭示了进化过程中TF结合特异性多样化的机制。 我们发现,三个不同结构TF家族的特定成员使用AT钩结构域来识别富含AT的序列,该序列位于TF及其旁系识别的基序两侧。 这种TF特异性多样化的机制与以前报道的类似( 26 )因此,在DBD附近添加精氨酸残基会导致优先选择与核心结合基序相邻的AAAA或TTTT序列。 总的来说,这些结果表明,TF特异性通常是通过添加简单的氨基酸特征来进化的,这些氨基酸特征使同源蛋白质识别的序列多样化。
我们的工作表明,最普遍的人类表观遗传DNA修饰,即CpG甲基化,对TF结合特异性的影响比以前认识到的更广泛。 与大多数先前报告的病例相比[例如( 9 , 10 , 12 , 17 , 18 )],我们发现甲基化在许多情况下可以积极影响TF结合( 16 , 62 )]. 例如,早期的研究发现,只有4%的植物TF偏好胞嘧啶甲基化位点。 这种与我们结果的差异可以用四种机制来解释。 首先,一些差异是由以下事实造成的,即本研究和先前研究中获得的TF数据集并不完整,也不代表整个TF概要的随机样本。 其次,所有偏好人类mCpG的TF主要家族在植物中都不存在,包括典型同源结构域、POU和NFAT蛋白。 第三,植物甲基化重复元素,但很少调控序列。 第四,非CpG甲基化在植物中非常常见,O'Malley发现的许多TF 等 . ( 63 )研究受到了这种修改的影响。 我们在这里发现,大多数TF可以与甲基化改变TF结合亲和力的位点结合。然而,这些TF中的大多数也可以与不包含CpG二核苷酸的位点相结合。 因此,对同一因子作出反应的两个调控元件对DNA甲基化的敏感性可能不同。 此外,一些DNA序列,例如包含CGGAA子序列的一些基序,可以被甲基化影响的负(ETS)或正(NFAT)TF结合。 因此,甲基化可以作用于选择与目标序列结合的因子。 这种选择性效应会对转录产生重大影响,尤其是在一种因子充当激活剂而另一种因子作为阻遏剂的情况下。
我们对同源结构域的结构分析表明,mCpG是通过氨基酸和甲基胞嘧啶的5-甲基之间的直接疏水相互作用识别的。 鉴于胸腺嘧啶和甲基胞嘧啶的结构相似性及其5-甲基的位置相同,很难确定为什么TF对mCpG的偏好发生了变化。 这可能是胸腺嘧啶高亲和力识别的一个简单结果。 然而,一些研究结果表明,这可能不是全部原因。 例如,所有规范同源结构域都倾向于包含AATT序列的相似核心序列。 然而,它们对mCpG的偏好随着三个位置氨基酸残基疏水性的函数而变化。 此外,在后同源结构域蛋白中观察到对mCpG的最强烈偏好,该家族在脊椎动物谱系中已经扩大,全基因组DNA甲基化是必需的。 无论mCpG的特异性如何演变,我们的结果表明,无论在体外还是在体内,它都会影响TF与DNA的结合,导致TF的占有率和活性发生变化,这是基于其识别基序的甲基化。
在这项工作中,我们从多个TF家族中鉴定了许多对发育重要的TF,这些TF家族倾向于与mCpG结合,我们还确定了同源结构域蛋白对mCpG-不同偏好背后的分子机制。 能够与甲基化序列结合的TF对细胞重编程可能特别重要,因为CpG甲基化是细胞重编程的屏障( 14 , 15 ). 在这方面,特别令人感兴趣的是,调节ES细胞自我更新的几个因子,包括PRDM4、Nanog和POU同源域因子POU5F1(OCT4),能够与mCpG位点结合。 这可能在一定程度上解释了POU5F1对分化细胞进行多能性命运重编程的能力( 48 ). 我们发现,许多TF更喜欢甲基化CpG,以及我们所产生的TF结合特异性的基因组规模资源,将对未来的表观遗传和转录调控分析具有重要意义。
材料和方法 克隆、蛋白质表达和纯化 以pETG20A质粒为骨架,构建了含有N末端硫氧还蛋白-6×His标签、C末端链亲和素结合肽(SBP)或3×FLAG标签的细菌蛋白表达网关受体载体。 通过哺乳动物基因收集(MGC)、ORFeome、Megaman cDNA文库中的聚合酶链反应(PCR)或基因合成(Genscript;参见 表S1 蛋白质序列和结构域),或来自先前发布的Gateway供体克隆( 21 ).
蛋白质表达和纯化 大肠杆菌 细胞按中所述进行( 64 ),具有以下修改:30µM ZnSO 4 被包括在培养基中以促进锌指蛋白的表达和折叠。 通过SDS-PAGE电泳(E-PAGE蛋白凝胶,Invitrogen)和考马斯亮蓝染色检查纯化蛋白的表达。 在−20°C储存之前,向蛋白质中添加50%的甘油。 结果比较表明,使用这种重组细菌蛋白恢复的基序高度相似( 图S1 和 表S2 )在培养的人类细胞中表达TF的实验中( 21 , 26 ).
HT-SELEX分析 HT-SELEX基本上按照Nitta所述执行 等 . ( 25 ). 简言之,由40 bp随机序列和Illumina测序适配器组成的选择配体由单链模板引物延伸生成。 然后将配体(循环0时约1.5µg,之后约200 ng)与标记纯化的六组氨酸硫氧还蛋白孵育 大肠杆菌 重组蛋白(100至200 ng)在微孔板中,在聚dI:dC竞争物(75 ng)存在下,然后通过镍亲和珠回收蛋白质和结合DNA。 该过程重复了四次,配体通过PCR扩增,并在每个循环后测序。 反应在含有较低盐浓度的缓冲液中进行(4%甘油、1 mM DTT、500µM EDTA、10 mM Tris-Cl中50 mM NaCl,pH 7.5) 在SELEX清洗步骤中,为了防止TF与DNA过度分离(低盐加强了TF和DNA骨架之间的非顺序特异性离子相互作用),在细胞核内发现的TF。 获得的基序与我们使用生理水平KCl(140 mM; 图S19B ).
最初的DNA数量足以包含所有20 bp的缺口和未缺口序列的大多数。 虽然由于DNA数量的限制,并非所有40 bp序列都被查询,但TF通常不会与40 bp(80位信息)的精确匹配相结合; 通过使用DNA的数量,HT-SELEX可以识别信息含量约为40位的基序,超过了大多数人类TF的信息含量[约15位( 26 )]. 有关HT-SELEX方法和分析的更多详细信息,请参阅( 21 , 26 ).
在SELEX中,由于TF对高亲和力位点的饱和,第一个周期低估了亲和力,随后的周期产生了高亲和力部位的指数富集。 在晚期周期(>4)中,大多数序列将包含一个与TF结合的序列,高亲和力序列将开始竞争中等亲和力位点,最终产生极少的单个序列。 我们之前已经将PBM和HT-SELEX与更直接测量(相对)亲和力的方法进行了比较,并且所获得的基序与PBM和早期周期(2或3)SELEX数据非常相似( 21 , 26 , 33 ). 因此,在这里使用相对早期的SELEX循环生成图案(在 表S2 ). 此外,由于相似性和丰富性的等级相同,精确的相似性对于使用阈值进行基序匹配的方法来说并不重要。 然而,从SELEX PWM获得的相对值只是亲和力的粗略估计,以及使用标准和/或方法(如Spec-seq)校准图案( 65 )如果需要精确的相对亲和力值,则应使用。
甲基-SELEX 甲基SELEX工艺基于HT-SELEX( 21 , 26 )在每个选择周期之前添加DNA甲基化步骤。 CpG甲基化由CpG特异性甲基化酶M.SssI执行。 甲基化协议改编自( 11 )如下:将2.5µl(10U,用于初始文库)或1.25µl(5U,用于周期1-3)CpG甲基转移酶M.SssI(NEB;在周期0中过量2倍,此后过量10倍)与0.4µl(用于初始文库)或0.2µl(用于周期1-3)S-腺苷甲硫氨酸、3.4µl 50mM氯化镁一起添加到DNA配体中 2 和0.2µl 100 mM DTT,总体积为20µl。 将混合物在37℃下培养3小时,使双链DNA中的CpG二核苷酸甲基化,然后在65℃下培养20分钟,在每个选择周期之前使M.SssI酶失活。 通过甲基化特异性限制酶BstBI检测配体的甲基化状态来优化甲基化反应( 图S19A ). 此外,在筛选中,在每个板中包括对照甲基特异性TF(HOXB13和/或ATF)。
CpG甲基化DNA配体和非甲基化DNA配体在不同的96 well板中对每个蛋白质进行平行的SELEX分析,选择过程重复4个周期以丰富结合序列,如( 21 , 26 ). 对所有四个周期的富集寡核苷酸进行测序。
探讨盐浓度对疏水相互作用的潜在影响,这可能会影响TF对mCpG的偏好( 66 ),我们还进行了140 mM KCl存在下的对照实验。 较高盐浓度的实验成功率较低,但获得的基序与使用50 mM NaCl缓冲液获得的基模没有实质性差异( 图S19B ).
亚硫酸氢盐-SELEX 甲基-SELEX可用于对与包含和不包含CpG的两个位点都具有某种亲和力的TF进行分类。 在甲基化块与最高亲和性位点结合的情况下,不含CpG的低亲和性序列比没有甲基化的序列更丰富。 含CpG基序的相对缺失将表明TF为甲基-微量。 然而,仅能与含CpG序列结合的TF很难分类,因为弱结合仍会产生一个基序,甚至被mCpG完全阻断的TF也可能由于M.SssI对DNA的不完全甲基化而在甲基-SELEX中产生更弱富集的含CpG-基序。 为此,我们开发了亚硫酸氢盐-SELEX,它允许在单轮SELEX中分析CpG甲基化的定量效应。 在该分析中,直接测量mCpG状态,因此M.SssI对DNA的部分甲基化不会影响结果。 然而,检测中可能存在一些半甲基化DNA。 使用亚硫酸氢盐-SELEX可以专门分析半甲基化DNA,因为该基序仅由一条DNA链生成; 然而,半甲基化分析需要生成半甲基化DNA,例如在甲基化后执行一个PCR循环。 由于根据结构分析,半甲基化的影响预计介于完全甲基化和非甲基化状态之间,因此未在此处进行分析。
在亚硫酸氢盐-SELEX中,混合的HT-SELEX和甲基-SELEX-富集的选择配体部分甲基化,并经受另一轮SELEX。 然后使用标准和亚硫酸氢盐测序对附加SELEX轮中选择的输入混合物和配体进行分析,以确定每个TF对甲基化CpG亚序列的偏好。
对于亚硫酸氢盐-SELEX,在其结合位点中具有CpG亚序列的蛋白质,在第3周期之前执行HT-SELEX和甲基-SELEX-过程。 然后将来自CpG甲基化和非甲基化DNA配体的富集DNA寡核苷酸混合在一起,然后一半的混合寡核苷酸进行如上所述的甲基化过程,然后与非甲基化寡核苷酸混合。 随后对CpG甲基化和非甲基化寡聚物的混合物进行额外的SELEX过程循环(循环4),并在70μl微Q水中洗脱富集寡聚体。 通过PCR扩增13μl洗脱液(Phusion DNA聚合酶,Fisher Scientific;65°C 10 s,72°C 36 s,97°C 15 s,分别用于退火、延伸和变性,20个循环),并通过qPCR(Roche LightCycler 480)分析3μl等分试样,以监测实验进展。 随后,对40µl洗脱液和5µl来自循环3的CpG甲基化和非甲基化寡聚物混合物进行亚硫酸氢盐处理(EZ-96 DNA甲基化金试剂盒,ZYMO RESEARCH)并通过PCR扩增(PfuTurbo Cx Hotstart DNA聚合酶,安捷伦科技公司; 前2个循环分别为60℃30 s、72℃60 s、95℃30 s,用于退火、伸长率和变性,随后的13或25个循环分别是65℃30 s和72℃60秒、95℃30s)。 从正常洗脱和亚硫酸氢钠处理洗脱扩增的第4周期PCR产物、CpG甲基化和非甲基化寡聚物混合物以及从第3周期亚硫酸氢处理的CpG甲甲基化和未甲基化寡核苷酸混合物中扩增的PCR产物均进行了测序。
为了计算模体中mCpG增加的百分比,根据与亚硫酸氢盐SELEX种子完全匹配的子序列确定特定位置的二核苷酸频率( 表S3 )除了被询问的二核苷酸位置以外的所有其他位置。 对于位置(以粗体显示 表S3 )如果甲基-SELEX或HT-SELEX-循环3的CpG计数高于10%,则mCpG频率从循环3到循环4的增加计算如下: (f) imCG公司 = ( (f) nmCG_周期4 / (f) nmCG_周期3 − 1) ×100%. 对于循环3和4,甲基化的归一化频率 (f) 纳米CG 根据以下方程式计算得出: (f) nmCG公司 = 平方英尺 × (f) 微CG ,其中尺寸系数 平方英尺 = 1/( (f) umCG公司 + (f) 微CG +假计数)和非甲基化CG的频率 (f) umCG公司 = [( (f) YG公司 − (f) TG公司 ) + ( (f) CG公司 − (f) 微CG )]/2; YG是亚硫酸氢盐处理后的TG计数(umCG和TG),假计数为10 −9 包括在内以避免被零除。
PWM模型的生成 如前所述,使用Autoseed管道分析来自非甲基化和甲基化配体的每个单独TF的结合模型( 25 , 64 ). 简单地说,计算了8个和10个bp的未映射子序列以及中间包含间隙的子序列,并使用“Huddinge距离”度量分析了它们之间的相似性( 25 ). Huddinge距离定义为 d日 − 一 ,其中 d日 是两个比较子序列中任意一个子序列中定义的最大基数,以及 一 是在不引入新间隙的情况下,它们之间可以完美对齐的最大基数。 每个TF的初始种子是使用局部最大子序列生成的(在Huddinge距离为1时,子序列的计数高于其任何相邻子序列)。 使用多项式方法使用这些种子生成初始PWM模型( 21 ),随后通过专家分析对种子进行了精炼[参见( 26 , 64 )]. 使用的精确种子、SELEX循环和多项式模型如所示 表S2 .
TF分类 根据亚硫酸氢盐-SELEX,将每个TF分为甲基+、甲基-微量、轻微影响或多重影响类别( 图5A 和 表S3 )除亚硫酸氢盐实验富集度低、种子复杂度低或无数据的情况外; 在这些情况下,未考虑亚硫酸氢盐-SELEX数据,分类基于甲基-SELEX。 在23个案例中,亚硫酸氢盐-SELEX和甲基-SLEX的数据或两个亚硫酸氢-SELEX实验之间的重复数据不一致,未分类,并在 表S3 首先,将每个基序分类为多重效应,甲基加、甲基减、小效应或无CpG类。 然后,将具有单个基序的TF指定为其基序类别,并将具有多个基序的TF分类如下:如果TF在多效应类别中具有任何基序, 或两个或多个对CpG甲基化表现出不同影响的基序(不同的基序属于以下两个或两个以上类别:甲基+、甲基-微量或小影响),它被归类为多重影响。 如果TF的基序没有CpG,而基序有CpG(s),则根据带有CpG的基序进行分类(参见 数据S2 详细信息)。
此外,甲基-plus和甲基-minus TF被分为A型或B型,以表明是否分别影响了最富集的位点或中等富集的位点(低于最大值但大于最大值的10%)。 对于每个TF,这种分类基于从其主要基序(出现次数最多的基序)衍生的一致序列。 使用基于一致序列的方法代替特定长度的kmer,因为不同TF的基序具有不同的长度,并且许多单个TF也丰富了两个或多个不同长度的基序。 如果一致序列包含CpG,则TF被分类为a型,如果没有,则被分类为B型。甲基-plus和甲基-minus TF分别基于使用甲基化和非甲基化配体富集的基序进行分类。
蛋白质结合微阵列 对于蛋白结合微阵列分析,使用网关LR反应将DLX3、POU5F1、MAX、NFATC2、CUX1和CUX2的DBD克隆转移到带有N末端GST标记的pDEST15。 从pDONR克隆中扩增出Nkx2.5的DBD并克隆到pETGEXCT的NcoI-SacI限制位点( 67 ). 带有N末端GST标记的LHX9的DBD来自Dr。 蒂莫西·休斯实验室(多伦多大学)。 所有克隆均经过序列验证。
16×HK阵列设计,具有40000个独特的DNA特征[如所述( 11 , 35 )]如中所述的双绞线( 11 ). 使用10μl CpG甲基转移酶M.SssI(20个单位/μl)(NEB)、1μl S-腺苷蛋氨酸和15μl 10×NEB缓冲液2在双链阵列上对CpG二核苷酸进行甲基化。 用0.005%Triton X-100将反应体积调整为150µl,并在37°C下培养3小时。 添加Triton X-100对阵列的完全甲基化至关重要。
蛋白质结合反应按照( 35 ). 简单地说,用4%脱脂奶粉在1×PBS(Sigma)中封闭双链微阵列1小时。 然后用0.1%(vol/vol)吐温-20的PBS清洗一次微阵列5分钟,用0.01%Triton X-100的PBS洗涤一次2分钟。根据制造商说明,使用PURExpress体外蛋白合成试剂盒(NEB)表达带有GST标记的DBD。 添加25μl IVT反应,使总体积为150μl的含PBS的蛋白质结合反应与2%(wt/vol)牛奶、51.3 ng/μl鲑鱼睾丸DNA(Sigma)和0.2μg/μl牛血清白蛋白(NEB)发生,并在20°C下孵育1小时。 将预孵育的蛋白结合混合物应用于40K阵列的单个腔室,并在20°C下孵育1小时。 在Coplin罐中用0.5%(vol/vol)吐温-20在PBS中清洗一次微阵列3分钟,用0.01%Triton X-100在PBS内清洗一次2分钟,最后用PBS清洗。将Alexa Fluor 647-共轭GST抗体(Invitrogen)涂敷到每个室中,并在20°C下培养1小时。 最后,用含有0.05%(vol/vol)吐温-20的PBS清洗微阵列两次,每次3分钟,在PBS中清洗一次,每次2分钟。 扫描蛋白结合微阵列以在640nm处检测Alexa Fluor 647缀合的抗GST。 使用ImaGene(BioDiscovery)分析微阵列图像,并将提取的数据用于进一步分析(GEO: GSE94634标准 ). 为了估计每种8-mer的相对偏好,Z评分是根据包含每种8-mer的16或32个点的平均信号强度计算的( 11 ).
细胞培养、细胞转导、ChIP-seq和ChIP-exo 泰特 -TKO(缺乏 测试1 , 测试2 、和 测试3 )其生成方式和遗传背景与 Dnmt公司 -TKO线( 51 )经过以下修改:在选择嘌呤霉素后,克隆最初根据前面描述的扩增和RFLP消化进行基因分型( 68 )使用以下引物和限制性内切酶组合: 测试1 (TTGTTCTCTCCTGACTGC、TGATTGATCAAATAGGCCTGC、SacI), 测试2 (CAGATGCTTAGGCCAATCAAG、AGAGCACACATGAAGATG、EcoRV), 测试3 (CCACCTCTGAGCGCAGAGTG、GATGAACAGTTCTGACAG、XhoI)。 这些研究中使用的阳性克隆在 测试1 ,8 bp和10 bp缺失 测试2 和8 bp纯合缺失 测试3 .
野生型, Dnmt公司 -TKO公司( 51 )(缺乏 Dnmt1型 , Dnmt3a型 、和 Dnmt3b型 )和 泰特 -将TKO细胞无饲养细胞培养在DMEM中的0.2%明胶涂层培养皿上,补充15%胎牛血清、1×非必需氨基酸、2mM L-谷氨酰胺、LIF和0.001%β-巯基乙醇(37℃,7%CO) 2 ) ( 51 ).
表达野生型雄激素受体(LSH-AR)的永生人前列腺上皮细胞是William Hahn教授(波士顿达纳-法伯癌症研究所)赠送的礼物。 细胞在PrEBM前列腺上皮基底培养基(Lonza)中培养,并添加生长因子补充剂(PrEGM SingleQuots(Lonsa)),并按上述方法传代( 69 ).
利用网关重组系统将全长HOXB13 ORF克隆到pLenti6/V5慢病毒表达载体中。 使用Lipofectamine 2000(Thermo Fisher Scientific)将表达载体与包装载体psPAX2和pMD2.G(Addgene)联合转染到293FT细胞中,从而产生病毒。 第二天,用新鲜的培养基补充细胞,48小时后收集含病毒的培养基。 使用Lenti-X浓缩器(Clontech)浓缩病毒。 在存在8µg/ml聚brene的情况下进行转导。 转导后16小时,用新鲜培养基替换LSH-AR细胞培养基,并进一步培养48小时。
染色质免疫沉淀法(ChIP)如前所述进行,但有轻微修改( 70 )通过在野生型和缺陷ES细胞中使用OCT4、KLF4、n-Myc抗体(Abcam cat.no.ab19857和R&D Systems cat.no.AF1759、AF3158和AF3640,以及Abcam cat.no.ab16898),或在转染的LHSAR细胞中使用HOXB13抗体。 简单地说,在室温下将细胞固定在1%甲醛中10分钟,然后添加0.125 M甘氨酸。 用冰镇PBS清洗细胞两次,并将其收集在裂解缓冲液中(5 mM PIPES,pH 8.0,85 mM KCl和0.5%NP-40)。 将细胞悬浮液离心,并将颗粒重新悬浮在含有蛋白酶抑制剂(Roche)的300µl RIPA缓冲液(1%NP‐40,0.5%脱氧胆酸钠,1×PBS中0.1%十二烷基硫酸钠)中进行溶解。 使用Bioruptor(Diagenode)将染色质超声至平均片段大小100–300 bp,然后在+4℃下以13000 rpm离心样品15 min,以收集上清液。 将Dynal蛋白-G磁珠(Invitrogen)用5mg/ml BSA在PBS中预洗涤总共5次,并重悬于100μl洗涤缓冲液中。 将感兴趣蛋白的特异性抗体与磁珠偶联过夜,并在+4 C下旋转。对于每个免疫沉淀(IP),用900µl RIPA缓冲液将100µl的超声染色质按1:10稀释,并将10%作为输入部分保存。 在剩余的样品中,添加100µl抗体偶联磁珠,并在4°C的旋转器上培养过夜。 培养后,用LiCl洗涤缓冲液(100 mM Tris-Cl,pH 7.5,500 mM LiCl,1%NP-40和1%脱氧胆酸钠)洗涤珠子5次,然后用含有1 mM EDTA的10 mM Tris-Cl(pH 8.0)洗涤两次。 在65°C的IP洗脱缓冲液(1%SDS,0.1 M NaHCO)中孵育1小时,从珠子中洗脱染色质抗体样品 三 ,在10mM Tris-Cl中,pH 7.5),然后在65°C下孵育过夜以反向交联。 使用酚氯仿纯化洗脱的DNA,然后准备文库进行Illumina测序。
LoVo(ATCC,cat.no.CCL229TM)细胞在添加10%胎牛血清(FBS)和抗生素的DMEM中培养。 当汇流达到60-70%时,ChIP-exo实验基本上按照Rhee和Pugh的描述进行( 71 )经过Katainen的修改 等 . ( 43 )通过使用CEBPB和MAX(以及圣克鲁斯生物技术分类号sc-150 X、sc-197和细胞信号技术分类号4732S)的抗体。 LoVo细胞KLF5的ChIP-exo数据来自Katainen 等 . ( 43 ).
使用bwa将ChIP文库中的原始测序读数映射到人类参考基因组(hg19)或小鼠参考基因组(mm9)( 72 )使用默认参数。 对于ChIP-exo峰值调用,我们使用带有默认参数的Peakzilla( 73 ). 对于ChIP-seq峰值呼叫,我们使用MACS(v1.4)( 74 )具有以下参数:未调整 P(P) < 10 −5 ; IgG控制的折叠转换≥2; 错误发现率≤5%。 LoVo、GP5D和VCaP细胞的ChIP-seq数据来自Yan 等 . ( 27 )和黄 等 . ( 75 ). 使用UCSC liftOver将峰值呼叫从hg18坐标转换为hg19坐标。 请参见 表S1 Illumina测序适配器的序列,以及 表S4 MEME检测到的峰区富集基序的对齐读取数和E值( 76 )每个实验。 使用BEDtools(v2.24.0)计算峰值重叠,要求重叠至少20%。 只要可用,重叠峰用于下游分析( 表S4 ).
为了确定被占据的区域是否在体内被甲基化,我们首先确定了ChIP-exo/seq峰区域和侧翼区域内的最佳得分基序匹配,这些区域距离任一方向上每个峰的边界都大于1kb但小于11kb, 然后测定含有CpG二核苷酸的基序匹配物中甲基化胞嘧啶的分数。 从HT-SELEX中富集的基序用于甲基-微量和小效应类别中的TF,从甲基-HT-SELEX中富集的模序用于甲基-plus组中的TFs。 使用程序MOODS从人类或小鼠基因组中搜索每个基序识别的得分最高的300000个位点( 77 )带有 P(P) 值截止值为10 −4 得分截止值为5。 从产生ChIP-exo/seq数据的各个细胞的全基因组亚硫酸氢盐测序(WGBS)数据中获得峰值和侧翼区域内最佳评分基序匹配的CpG子序列中的胞嘧啶甲基化。 然后使用R直方图比较胞嘧啶甲基化百分比在峰和侧翼区域的分布。 包含了CpG子序列中的所有胞嘧啶,其具有≥2的读取覆盖率的最佳评分基序匹配。 然而,结果对截止值的变化是稳健的( 数据S3 ). OCT4和n-Myc峰的基序匹配来自 泰特 和 Dnmt公司 -合并TKO ChIP-seq数据,并使用BEDtools genomecov(v2.26.0),根据库片段长度延伸的ChIP-se序列读取计算峰值高度。 两个细胞的两个ChIP-seq实验的平均片段覆盖率表示为 图6A 和 图S13D , S14B型 、和 S14C系列 .
测序和亚硫酸氢盐测序 使用PCR-纯化试剂盒(Qiagen)纯化未选择和选择的SELEX文库,并使用Illumina HiSeq 2000进行测序[复用400倍,否则与Jolma中的一样 等 . ( 21 ); 55 bp单读长度]。 原始序列读取被解复用,并使用Autoseed管道进行分析( 25 ). 每个周期和实验的读取计数范围约为100000到500000次读取。
来自GP5d、LoVo、VCaP、LHSAR、野生型和缺陷ES细胞的亚硫酸氢盐序列库是按照Illumina的说明准备的,只做了少量修改。 基因组DNA加入0.5%的非甲基化λDNA(Promega),并使用Covaris的200-bp靶峰大小协议在Covaris中进行片段化。对声波DNA样本进行末端修复、dA-tailed并连接到测序适配器。 使用ZYMO-EZ DNA甲基化金试剂盒处理连接的DNA片段以进行亚硫酸氢盐转化。 亚硫酸氢盐转化的DNA样本通过PCR(4至7个周期)进行富集。 使用Illumina HiSeq4000进行测序,使用100 bp配对读取,原始测序读取是高质量的,适配器使用Trim Galore中的cutadapt版本1.3进行修剪。 使用Phred得分截止值30进行低质量末端修剪。 适配器修剪是使用具有默认参数的标准Illumina双端适配器的前13 bp进行的。 使用Bismark(版本v0.10.0)对hg19或mm9参考基因组进行读取比对( 78 )和Bowtie 2(版本2.2.4)( 79 ). 使用Bismark重复数据消除功能删除重复数据。 甲基化调用的提取是通过Bismark甲基化提取器完成的,从第一次读取(–no_overlap参数)中丢弃配对读取重叠部分的前10 bp读取和读取甲基化调用。 GP5d、LoVo、VCaP、LHSAR、野生型和有缺陷ES细胞的WGBS覆盖范围的宽度和深度见 数据S3 。DSS检测到不同的甲基化区域( 80 ). 用于小鼠ES细胞的DNAse I超敏位点 图S13C 从UCSC的ENCODE数据(wgEncodeUwDnaseEscj7S129ME0PkRep1)下载。 窄峰),类似于Stadler之前的研究 等 . ( 8 )和热图是使用deepTools(2.4.1版)生成的( 81 ). 所有序列数据以注册号PRJEB9797保存在ENA(欧洲核苷酸档案馆)。
ES细胞的ATAC测序 使用转座酶可获得染色质的测序分析(ATAC-seq)捕获野生型和TKO ES细胞的开放染色质区域。 ATAC-seq基本上按照Buenrostro所述执行 等 . ( 82 )经过以下改进:通过胰蛋白酶法获取培养的细胞(70%融合),重新悬浮成单个细胞,用冰镇PBS洗涤并在500×g下离心5min。将细胞颗粒(50000个细胞)重新悬浮在2×溶解缓冲液(10mM NaCl,3mM MgCl)中 2 ,在10mM Tris-HCl中的0.1%Igepal CA-630,pH 7.5)和细胞核,通过在4°C下以500×g离心30分钟,使用具有低加速度和制动设置的摆动桶转子将其制成颗粒。 丢弃上清液,在25µl体积中对细胞核进行标记反应,其中含有2µl Tn5转座酶和12.5µl 2×TD缓冲液(来自Illumina的Nextera DNA Library Prep Kit,目录号FC-121–1030)。 在37°C的摇晃培养箱(650 rpm)中进行标记1小时。 标记后,添加5µl清洁缓冲液(900mMNaCl,300 mM EDTA)、2µl 5%SDS和2µl蛋白酶K(Thermo Fisher Scientific,目录号EO0491),并在40°C下进一步培养反应30分钟,剧烈摇晃(650 rpm)。 使用2×Agencourt AMPure XP SPRI珠(Beckman Coulter,目录号A63881)分离标记DNA,并使用22.5µl洗脱缓冲液(10 mM Tris-Cl,pH 8.0)洗脱DNA。 文库扩增分别使用6个周期和8个周期的两个连续PCR进行。 第一次PCR后,使用反相0.55X AMPure XP SPRI珠进行文库大小选择(对于小于800 bp的片段),收集上清液并使用MinElute PCR纯化试剂盒(QIAGEN,目录号28004)分离DNA。 使用2µl索引引物(来自Illumina的Nextera Index Kit,目录号FC-121–1011)和KAPA HiFi HotStart ReadyMix(KAPA Biosystems,KK2601)进行两次连续PCR(Illuminia的Nextra DNA Library Prep Reference Guide)。 使用Qubit 3.0荧光计(Thermo Fisher Scientific)测量最终DNA浓度,并使用2100生物分析仪(安捷伦科技公司)确定库大小。 根据制造商的说明,使用Illumina HiSeq 4000进行单基因55 bp测序。
使用bwa将原始测序读数映射到小鼠参考基因组(mm9)( 72 )使用默认参数。 使用Picard Tools MarkDuplicates去除重复,使用MACS2(2.0.9版)检测开放染色质区域( 74 )使用默认参数进行宽峰值调用。 ATAC-seq校准和峰值呼叫统计汇总于 表S4 两个重复的重叠峰(至少20%重叠)用于下游分析。
模体守恒与相似性分析 为了确定与基序的匹配是否被保存,根据( 64 )该程序在900个基序中检测到598个基序(66.4%)的基因组保守性,家族错误率<0.05。 简单地说,每个基序的两万个顶级亲和位点是从人类限制的元素中选择的(不是全基因组,只有哺乳动物中的序列是保守的),并根据中解释的基序在99种脊椎动物物种中进行了保护检查(从UCSC基因组浏览器hg19版下载的多重比对)( 64 ). 为了将模体与保守结合位点的替换模式进行比较,我们选择了前千个亲和力最高的保守位点,或者如果少于千个位点,则选择所有保守位点进行进一步分析。 对于保守结合位点的每个位置,使用SiPhy程序(任务7)根据该位置的多重比对估计特定位置的平衡基分布π。 基分布π描述了作用于位置的进化约束,假设位置独立进化( 83 ). 守恒模式是通过平均保守位点每个位置的π频率来构建的。 修正获得的多重假设检验 P(P) 值(霍尔姆方法)和测试位点中保守基序位点的数量如所示 图S12 .
由于甲基化CpG的高突变负荷,这种分析往往低估了含CpG序列的保守性。 然而,为了避免测量变量依赖于校正项,我们没有校正突变率。 HT-SELEX图案和保守图案之间的差异标志是通过减去相应的基频来实现的。
使用SSTAT计算Motif相似性( 42 )使用严格的I型误差阈值0.01限制低亲和力位点的影响(其他参数50%GC背景模型,伪计数正则化)。 我们以前曾报道过,这种方法通常与其他常用方法产生类似的结果,但当两个不同的基序共享一个公共部分时,效果更好( 26 ). 如果SSTAT相似性得分>1.5×10,则将结合模型相互连接 −5 然后,使用所得网络的最小支配集来选择具有代表性的PWM( 25 , 26 ). 最小支配集是在相似网络中直接连接到原始集的所有PWM的最小PWM集。
富集分析 通过考虑TFs的集合进行富集分析,我们获得了一个基序作为参考群体,以避免采样偏差。 由于SELEX富集是使用甲基化和非甲基化配体进行的,我们不认为这两类配体之间的种群会有特定的偏差。 使用PANTHER进行GO富集分析( 84 )过度陈述测试(2016年7月15日发布),注释版本11.1。
结构分析和等温滴定量热法 人LHX4(残基153–220)、HOXB13(残基209–283)、MEIS1(残基279–333)、CDX1(残基154–217)和CDX2(残基169–262)的DNA结合域片段的表达和纯化如( 85 ). 结晶中使用的DNA片段以单链寡核苷酸(Eurofins MWG)的形式获得,并在含有150 mM NaCl、1 mM三(2-羧乙基)膦(TCEP)和5%甘油的20 mM HEPES(pH 7.5)中退火。 首先将纯化和浓缩的蛋白质与退火DNA双链溶液混合,LHX的摩尔比为2:1.2,HOXB13、CDX1和CDX2的摩尔比是1:1.2,HOXB13:MEIS1复合物的摩尔比则是1:1:1.2,然后在冰上放置1小时后进行结晶试验。 使用Jena Bioscience JBScreen Nuc-Pro HTS(Jena Bios science,Jena,Germany)优化结晶条件。 所有蛋白-DNA复合物在室温下通过蒸汽扩散技术在坐滴液中结晶。 LHX4-DNA从含有50 mM Tris缓冲液(pH 8.0)、40 mM甲酸镁、30%(w/v)聚乙二醇单甲醚[PEGmme(5000)]和4%2-甲基-2,4-戊二醇(MPD)的溶液中结晶。 HOXB13-MethDNA复合物从含有50 mM Tris缓冲液(pH 8.0)、100 mM MgCl的溶液中结晶 2 ,150 mM KCl,24%(w/v)PEG(3350)和8%甲基丙醇。 从含有50 mM Tris缓冲液(pH 8.0)、100 mM MgCl的溶液中结晶CDX1-MethDNA和CDX2-MethDNA复合物 2 ,150 mM KCl,28.8%(w/v)PEGmme(5000)和5%的MPD用于CDX1_methDNA或8%的聚乙二醇(400)用于CDX2_methDNA。 从含有50 mM Tris缓冲液(pH 8.0)、100 mM MgCl的溶液中生长了HOXB13、MEIS1与甲基化DNA的异二聚物晶体 2 ,150 mM KCl,24%(w/v)PEG(3350)和8%的1-戊醇。
所有数据集都是在欧洲同步辐射设施(ESRF,法国格勒诺布尔)收集的,在100 K时,使用储存溶液作为防冻剂,从束线ID29(LHX4和HOXB13:MEIS1_methDNA)和ID23–1(HOXB13_methDNA、CDX1_methDNA和CDX2_meth脱氧核糖核酸)上的单晶收集。 使用BEST程序优化数据收集策略( 86 ). 数据与程序XDS集成( 87 )并使用SCALA进行缩放( 88 ). 数据收集统计数据见 表S6 .
使用Phaser程序通过分子替换技术求解所有结构( 89 )在凤凰城实施( 90 )和CCP4( 88 )和CCP4程序套件下的Molrep。 首先利用MEIS1(PDB条目4XRM)结构作为搜索模型的Phaser程序,通过分子替换求解HOXB13:MEIS1_methDNA的结构。 所找到的MEIS1的解决方案是固定的,并且使用来自HOXA9/PBX1复合体的HOXA9的坐标(PDB条目1PUF)来确定HOXB13的位置。 两种蛋白质定位后,DNA密度清晰,并使用COOT手动构建分子( 91 , 92 ). 使用REFMAC5进行刚体细化,然后使用REFMAC进行约束细化,如CCP4和Phenix refine中所实现的那样( 93 ). HOXB13的精细模型后来被用作搜索模型,以确定HOXB12与甲基化DNA的结构,并用于求解CDX1_methDNA和CDX2_methDNA结构(序列一致性为43%)。 DNA与蛋白质相互作用的部分清晰可见,并手动构建到出现的电子密度并进行精细化。
LHX4的搜索模型是大鼠胰岛素基因增强子蛋白ISL-1的同源结构域,作为搜索模型,该同源结构域与LHX4(PDB条目1BW5)的DBD具有48%的一致性。 LHX4-DNA复合物第二亚单位的电子密度在这个阶段不太明显。 包含LHX4和部分DNA的结果模型是固定的,第二个LHX4分子的位置由程序Molrep使用实现的球形平均相转移函数(SAPTF)确定,如( 94 ). 两个蛋白质分子定位后,DNA密度清晰,DNA分子使用COOT手动构建( 91 , 92 ).
使用Phenix对包含一个DNA和两个蛋白质分子的结果模型进行了改进。 提炼( 93 )使用TLS功能。 由于缺乏填料接触,第二亚基的密度远低于第一亚基的呈现密度。 第一亚基N端和C端的前3个和最后4个氨基酸以及第二亚基N末端的7个第一氨基酸和C末端的4个最后氨基酸被发现是无序的,没有被纳入最终模型。 细化统计信息如所示 表S6 实验数据和原子坐标已提交给蛋白质数据库,加入代码为5HOD(LHX4)、5EF6(HOXB13_methDNA)、5EGO(HOXB1 3:MEIS1_methDNA),5LUX(CDX1_methDNA)和5LTX(CDX2_meth脱氧核糖核酸)。
为了确定ONECUT2、LHX4、HOXA11和HOXB13 DBD对各自甲基化和非甲基化DNA基序的亲和力,使用ITC200微量热量计(美国马萨诸塞州北安普敦MicroCal)进行了等温滴定量热实验 PSF(瑞典卡罗林斯卡研究所蛋白质科学设施和瑞典GE Healthcare)。 通过直接滴定蛋白质到含有指示双链DNA配体的细胞,测定DNA的结合等温线。 在20°C下进行测量。 蛋白质和DNA均在含有20mM HEPES pH7.5、300mM NaCl、10%甘油和0.5mM TCEP的缓冲液中制备。 共进行20次注射,每次注射间隔240秒。 所有数据均使用热量计附带的OriginPro 7.0软件包(Microcal)进行评估。 表观离解常数 K(K) d日 ,结合焓Δ H(H) -和化学计量 n个 以及它们相应的标准偏差,通过非线性最小二乘法将数据拟合到标准绑定方程中,使用软件包中实现的一组独立和相同的绑定位点的模型进行绑定。 结合熵和自由能由Δ关系得到 G公司 = − RT公司 自然对数 K(K) d日 =ΔH− T型 Δ S公司 .
荧光素酶分析 由EurofinsGenomics公司合成了含有8个HOXC11结合位点的寡核苷酸( 表S1 )利用Sbf I和Spe I限制性内切酶将其克隆到pCpGfree启动子报告质粒(InvivoGen)中,该质粒含有报告基因(分泌型荧光素酶Lucia),完全不含CpG二核苷酸。 利用CpG特异性甲基化酶M.SssI将重构的pCpGfree_promotor质粒中的胞嘧啶甲基化。 将含有甲基化或非甲基化CpG位点的质粒与含有HOXC11基因的pCDNA3.1–3×FLAG-V5表达载体(多伦多大学Mikko Taipale赠送)和含有 雷尼利亚 使用FuGENE HD转染试剂(Promega)的荧光素酶(Promeca)。 露西亚和 雷尼利亚 36小时后,通过双Glo萤光素酶测定系统(Promega)和EnVision多标记读取器(PerkinElmer)测量萤光素酶活性。
致谢 我们感谢S.Linnarsson、N.Zielke、B.Schmierer、F.Zhu和J.Zhang对手稿的批判性审查; S.Augsten和卡罗林斯卡研究所蛋白质科学设施,用于蛋白质生产和运行ITC实验的机会; 以及L.Hu、A.Zetterlund和M.Hoh提供技术援助。 Illumina测序读数保存在欧洲核苷酸档案馆,登记号为PRJEB9797。 实验数据和原子坐标已提交给蛋白质数据库,加入代码为5HOD(LHX4)、5EF6(HOXB13_methDNA)、5EGO(HOXB13:MEIS1_methDNA),5LUX(CDX1_methDNA)和5LTX(CDX2_meth脱氧核糖核酸)。 这项工作得到了芬兰癌症遗传学研究院卓越中心、欧洲研究区SynBio项目MirrorBio、卡罗林斯卡研究所创新医学中心、克努特和爱丽丝·沃伦伯格基金会、戈兰·古斯塔夫森基金会和Vetenskapsr-det的支持。 Y.Y.、A.J.、K.R.N.和J.T.设计了实验。 Y.Y.进行了HT-SELEX、甲基-SELEX-和亚硫酸氢盐-SELEX.实验。 Y.Y.和B.S.执行ChIP-seq实验,K.D.执行ChIP-exo和ATAC-seq实验,B.S.进行全基因组亚硫酸氢盐测序实验。 S.K.S.进行了蛋白结合微阵列实验。 P.K.D.纯化的蛋白质用于结晶学,E.M.和A.P.进行了x射线结晶学实验,而E.M.解决了蛋白质结构。 P.A.G.和S.D.生成 泰特 -TKO ES细胞。 Y.Y.、E.K.、K.R.N.、A.J.、F.Z.、T.K.和J.T.对计算分析做出了贡献。 Y.Y.、E.K.和K.R.N.准备了插图。 Y.Y.和J.T.写了手稿。 所有作者都参与了数据分析并审阅了手稿。
参考文件和注释
1 Huff JT、Zilberman D、Dnmt1-依赖性CG甲基化有助于核小体在不同真核生物中的定位。 单元格 156, 1286–1297 (2014). doi:10.1016/j.cell.2014.01.029 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
2 凯利TK 等人,个体DNA分子内核小体定位和DNA甲基化的全基因组图谱。 基因组研究 22, 2497–2506 (2012). doi:10.1101/gr.143008.112 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
三。 鸟A,DNA甲基化模式和表观遗传记忆。 基因开发 16, 6–21 (2002). doi:10.1101/gad.947102 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
4 球MP 等,靶向和基因组规模策略揭示了人类细胞中的基因体甲基化特征。 自然生物技术 27, 361–368 (2009). doi:10.1038/nbt.1533 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
5 李斯特R 等,碱基分辨率的人类DNA甲基体显示出广泛的表观基因组差异。 自然 462, 315–322 (2009). doi:10.1038/nature08514 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
6 GC议员 等,在成年小鼠组织的DNA甲基化图谱中确定的胚胎增强因子的表观遗传记忆。 自然遗传学 45, 1198–1206 (2013). 数字对象标识代码:10.1038/ng.2746 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
7 Schübeler D,DNA甲基化的功能和信息含量。 自然 517, 321–326 (2015). doi:10.1038/nature14192 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
8 Stadler MB(Stadler MB) 等,DNA-结合因子在远端调节区塑造小鼠甲基体。 自然 480, 490–495 (2011). doi:10.1038/nature10716 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
9 Gaston K、Fried M、CpG甲基化对YY1和ETS蛋白与Surf-1和Surf-2基因双向启动子的结合有不同的影响。 核酸研究 23, 901–909 (1995). doi:10.1093/nar/23.6901 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
10 Iguchi-Ariga SM,Schaffner W,cAMP应答增强子/启动子序列TGACGTCA的CpG甲基化消除特异性因子结合和转录激活。 基因开发 3, 612–619 (1989). doi:10.1101/gad.3.5.612 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
11 Mann IK公司 等,CG甲基化微阵列确定了一个新的甲基化序列,该序列与体内活性的CEBPB | ATF4异二聚体结合。 基因组研究 23, 988–997 (2013). doi:10.101/克146654.112 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
12 Watt F,Molloy PL,胞嘧啶甲基化阻止与HeLa细胞转录因子的DNA结合,该转录因子是腺病毒主要晚期启动子最佳表达所必需的。 基因开发 2, 1136–1143 (1988). 数字对象标识代码:10.1101/gad.2.9.1136 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
13 Klose RJ,Bird AP,基因组DNA甲基化:标记及其介体。 生物化学趋势。 科学 31, 89–97 (2006). doi:10.1016/j.tibs.2005.12.008 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
14 鲁尼·TJ 等,通过细胞融合对闭塞基因的系统定位揭示了顺式介导的沉默在体细胞中的普遍性和稳定性。 基因组研究 24, 267–280 (2014). doi:10.1101/gr.143891.112 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
15 马球JM 等,将体细胞重新编程为iPS细胞的分子路线图。 单元格 151, 1617–1632 (2012). doi:10.1016/j.cell.2012.11.039 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
16 里希五世 等,半CRE序列的CpG甲基化产生C/EBPα结合位点,激活一些组织特异性基因。 程序。 国家。 阿卡德。 科学。 美国 107, 20311–20316 (2010). doi:10.1073/pnas.1008688107 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
17 Campanero MR、Armstrong MI、Flemington EK、CpG甲基化作为E2F活性调节机制。 程序。 国家。 阿卡德。 科学。 美国 97, 6481–6486 (2000). doi:10.1073/pnas.100340697 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
18 Comb M,Goodman HM,CpG甲基化抑制前脑啡肽基因表达和转录因子AP-2的结合。 核酸研究 18, 3975–3982 (1990). doi:10.1093/nar/18.13.3975 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
19 胡S 等,DNA甲基化为人类转录因子提供了不同的结合位点。 电子生活 2,e00726(2013)。 doi:10.7554/eLife.00726 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
20 斯普鲁伊特CG 等,5-(羟基)甲基胞嘧啶及其氧化衍生物的动态读取器。 单元格 152, 1146–1159 (2013). doi:10.1016/j.cell.2013.02.004 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
21 乔尔玛A 等人,多重大规模平行SELEX,用于表征人类转录因子结合特异性。 基因组研究 20, 861–873 (2010). doi:10.10101克/100克552.109 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
22 Oliphant AR,Struhl K,通过随机选择确定大肠杆菌启动子元件的一致序列。 核酸研究 16, 7673–7683 (1988). doi:10.1093/nar/16.15.7673 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
23 Vaquerizas JM、Kummerfeld SK、Teichmann SA、Luscombe NM,《人类转录因子普查:功能、表达和进化》。 自然版本基因 10, 252–263 (2009). doi:10.1038/nrg2538 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
24 克洛泽RJ 等,MeCP2的DNA结合选择性,因为需要与甲基-CpG相邻的a/T序列。 摩尔细胞 19, 667–678 (2005). doi:10.1016/j.molcel.2005.07.021 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
25 尼塔KR 等,转录因子结合特异性在6亿年的双耳进化中的保护。 电子生活 4,e04837(2015)。 doi:10.7554/寿命.04837 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
26 乔尔玛A 等人,人类转录因子的DNA结合特异性。 单元格 152, 327–339 (2013). doi:10.1016/j.cell.2012.12.009 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
27. 严J 等,人类细胞中的转录因子结合发生在凝集素锚定位点周围形成的密集簇中。 单元格 154, 801–813 (2013). doi:10.1016/j.cell.2013.07.034 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
28 Meng X,Brodsky MH,Wolfe SA,用于确定转录因子的DNA结合特异性的细菌单杂交系统。 自然生物技术 23, 988–994 (2005). doi:10.1038/nbt1120 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
29 Orenstein Y,Shamir R,《从PBM、HT-SELEX和ChIP数据中获得的转录因子结合模型的比较分析》。 核酸研究 42,e63(2014)。 doi:10.1093/nar/gku117 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
30 Farh KK公司 等,因果性自身免疫病变体的遗传和表观遗传精细定位。 自然 518, 337–343 (2015). doi:10.1038/nature13835 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
31 法利EK 等,发育促进剂的亚优化。 科学类 350, 325–328 (2015). doi:10.1212/science.aac6948 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
32 维尔斯特拉J 等,小鼠调控DNA景观揭示了顺调控进化的全球原则。 科学类 346, 1007–1012 (2014). doi:10.1126/science.1246426 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
33 魏国浩 等,ETS家族DNA结合的体内外全基因组分析。 欧洲工商管理硕士J 29, 2147–2160 (2010). doi:10.1038/emboj.2010.106 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
34 Boyer LA、Latek RR、Peterson CL、The SANT domain:一个独特的组蛋白尾绑定模块? 自然修订分子细胞生物学 5, 158–163 (2004). 数字对象标识代码:10.1038/nrm1314 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
35. 巴迪斯G 等,转录因子识别DNA的多样性和复杂性。 科学类 324, 1720–1723 (2009). doi:10.1212/science.1162327 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
36 Hume MA、Barrera LA、Gisselbrecht SS、Bulyk ML、UniPROBE,2015年更新:关于蛋白质-DNA相互作用的蛋白质结合微阵列数据在线数据库的新工具和内容。 核酸研究 第43页,D117–D122(2015)。 doi:10.1093/nar/gku1045 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
37 Weirauch MT公司 等,真核转录因子序列特异性的测定和推断。 单元格 158, 1431–1443 (2014). doi:10.1016/j.cell.2014.08.009 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
38 Harrington MA、Jones PA、Imagawa M、Karin M,胞嘧啶甲基化不影响转录因子Sp1的结合。 程序。 国家。 阿卡德。 科学。 美国 85, 2066–2070 (1988). doi:10.1073/pnas.85.7.2066 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
39 Höller M,Westin G,Jiricny J,Schaffner W,Sp1转录因子结合DNA并激活转录,即使结合位点是CpG甲基化。 基因开发 2, 1127–1135 (1988). doi:10.1101/gad.2.9.1127 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
40 查特吉R 等,小鼠原代女性皮肤成纤维细胞和角质形成细胞的高分辨率全基因组DNA甲基化图。 表观遗传学染色质 7, 35 (2014). doi:10.1186/1756-8935-7-35 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
41 刘毅 等,Klf4识别甲基化DNA的结构基础。 核酸研究 42, 4859–4867 (2014). doi:10.1093/nar/gku134 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
42 Pape UJ,Rahmann S,Vingron M,位置频率矩阵之间的自然相似性度量及其在聚类中的应用。生物信息学 24, 350–357 (2008). doi:10.1093/bioinformatics/btm610 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
43 卡塔宁R 等,CTCF/凝集素结合位点在癌症中经常发生突变。 自然遗传学 47, 818–821 (2015). doi:10.1038/ng.3335 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
44 博加尼D 等,PR/SET域锌指蛋白Prdm4调节胚胎干细胞中的基因表达,但在发育中的小鼠胚胎中发挥非本质性作用。 分子细胞。 生物 33, 3936–3950 (2013). doi:10.1128/MCB.00498-13 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
45 Kmita M,Duboule D,组织时空轴; 25年的共线修补。科学 301, 331–333 (2003). doi:10.1212/科学1085753 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
46. 麦克马洪AP,神经模式:Nkx基因在脊髓腹侧的作用。 基因开发 14, 2261–2264 (2000). doi:10.1101/gad.840800 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
47 Pearson JC、Lemons D、McGinnis W,《动物身体模式中调节Hox基因功能》。 自然版本基因 6, 893–904 (2005). doi:10.1038/nrg1726 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
48 Takahashi K,Yamanaka S,通过特定因子从小鼠胚胎和成年成纤维细胞培养物中诱导多能干细胞。 单元格 126, 663–676 (2006). doi:10.1016/j.cell.2006.07.024 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
49 桥本H 等,Wilms肿瘤蛋白识别特定DNA序列中的5-羧基胞嘧啶。 基因开发 28, 2304–2313 (2014). doi:10.1101/gad.250746.114 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
50 王毅 等,WT1招募TET2来调节其靶基因表达并抑制白血病细胞增殖。 摩尔细胞 57, 662–673 (2015). doi:10.1016/j.molcel.2014.12.023 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
51 Domcke S公司 等,DNA甲基化和转录因子之间的竞争决定了NRF1的结合。 自然 528, 575–579 (2015). doi:10.1038/nature16462 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
52. Khund-Sayeed S号 等,E-box基序ACAT|GTG和ACAC|GTG中的5-羟甲基胞嘧啶增加B-HLH转录因子TCF4的DNA结合。 集成。 生物 8, 936–945 (2016). doi:10.1039/C6IB00079G [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
53 Yu M公司 等,哺乳动物基因组中5-羟甲基胞嘧啶的碱基再溶分析。 单元格 149, 1368–1380 (2012). doi:10.1016/j.cell.2012.04.027 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
54 波美兰茨MM 等,雄激素受体池在人类前列腺肿瘤发生中被广泛重组。 自然遗传学 47, 1346–1351 (2015). 数字对象标识代码:10.1038/ng.3419 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
55 Covde S,Abate-Shen C,Geiger JH,Msx-1同源结构域/DNA复合物的晶体结构。 生物化学 40, 12013–12021 (2001). doi:10.1021/bi0108148 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
56 乔希R 等,通过识别小凹槽结构介导的Hox蛋白的功能特异性。 单元格 131, 530–543 (2007). doi:10.1016/j.cell.2007.09.024 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
57 LaRonde-LeBlanc NA,Wolberger C,与DNA结合的HoxA9和Pbx1的结构:Hox六肽和前后DNA识别。 基因开发 17, 2060–2072 (2003). 数字对象标识代码:10.1101/gad.1103303 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
58 Passner JM、Ryoo HD、Shen L、Mann RS、Aggarwal AK,结合DNA的Ultrabithorax-Extradenticle同源结构域复合物的结构。 自然 397, 714–719 (1999). 数字对象标识代码:10.1038/17833 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
59 Piper DE,Batchelor AH,Chang CP,Cleary ML,Wolberger C,与DNA结合的HoxB1-Pbx1异二聚体的结构:六肽和第四同源结构域螺旋在复合物形成中的作用。 单元格 96, 587–597 (1999). doi:10.1016/S0092-8674(00)80662-5 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
60 Zhang Y,Larsen CA,Stadler HS,Ames JB,HOXA13序列特异性DNA结合和蛋白质二聚化的结构基础。 PLOS ONE系列 6,e23069(2011)。 doi:10.1371/journal.pone.0023069 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
61 Dragan人工智能 等,驱动同源结构域与DNA结合的力。 生物化学 45, 141–151 (2006). doi:10.1021/bi051705m [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
62 昆纳维尔S 等,在胚胎干细胞中,ZFP57/KAP1识别甲基化的六核苷酸以影响印迹控制区域的染色质和DNA甲基化。 摩尔细胞 44, 361–372 (2011). doi:10.1016/j.molcel.2011.08.032 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
63 奥马利RC 等,睫状体和外胚轴特征塑造了调控DNA的景观。 单元格 165, 1280–1292 (2016). doi:10.1016/j.cell.2016.04.038 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
64 乔尔玛A 等,转录因子对依赖DNA的形成改变了其结合特异性。 自然 527, 384–388 (2015). doi:10.1038/nature15518 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
65 Stormo GD,Zuo Z,Chang YK,Spec-seq:通过测序确定蛋白-DNA结合特异性。 简介。 功能。 基因组学 14, 30–38 (2015). doi:10.1093/bfgp/elu043 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
66. Khrapunov S公司 表观遗传调控蛋白MeCP2的DNA结合结构域的异常特征决定了其结合特异性。 生物化学 53, 3379–3391 (2014). doi:10.1021/bi500424z [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
67 Sharrocks AD,一种用谷胱甘肽S-转移酶产生N端和C端融合蛋白的T7表达载体。 基因 138, 105–108 (1994). doi:10.1016/0378-1119(94)90789-7 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
68 王H 等,通过CRISPR/Cas介导的基因组工程,一步生成携带多个基因突变的小鼠。细胞 153, 910–918 (2013). doi:10.1016/j.cell.2013.04.025 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
69 伯杰·R 等,雄激素诱导的人类前列腺上皮细胞分化和致瘤性。 癌症研究 64, 8867–8875 (2004). doi:10.1158/008-5472.CAN-04-2938 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
70 萨胡B 等,FoxA1在雄激素受体与染色质结合、雄激素信号传导和前列腺癌中的双重作用。 欧洲工商管理硕士J 30, 3962–3976 (2011). doi:10.1038/emboj.2011.328 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
71 Rhee HS,Pugh BF,在单核苷酸分辨率下检测到的全基因组蛋白质-DNA相互作用。 单元格 147, 1408–1419 (2011). doi:10.1016/j.cell.2011.11.013 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
72 Li H,Durbin R,使用Burrows-Wheeler变换进行快速准确的短读对齐。 生物信息学 25, 1754–1760 (2009). doi:10.1093/bioinformatics/btp324 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
73 Bardet AF,He Q,Zeitlinger J,Stark A,比较ChIP-seq分析的计算管道。 自然协议 7, 45–61 (2012). doi:10.1038/nprot.2011.420 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
74 张Y 等,基于模型的ChIP-Seq分析(MACS)。 基因组生物学 9,R137(2008)。 doi:10.1186/gb-2008-9-9-r137 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
75 黄Q 等,前列腺癌易感性等位基因6q22通过调节HOXB13染色质结合增加RFX6的表达。 自然遗传学 46, 126–135 (2014). doi:10.1038/ng.2862 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
76 Bailey TL、Williams N、Misleh C、Li WW、MEME:发现和分析DNA和蛋白质序列基序。 核酸研究 34,W369–W373(2006)。 doi:10.1093/nar/gkl198 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
77 Korhonen J,Martinmäki P,Pizzi C,Rastas P,Ukkonen E,MOODS:快速搜索DNA序列中的位置权重矩阵匹配。 生物信息学 25, 3181–3182 (2009). doi:10.1093/bioinformatics/btp554 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
78 Krueger F、Andrews SR、Bismark:一种适用于亚硫酸氢盐-Seq应用的灵活对准器和甲基化调用者。 生物信息学 27, 1571–1572 (2011). doi:10.1093/bioinformatics/btr167 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
79 Langmead B,Salzberg SL,与Bowtie 2进行快速定距对准。 自然方法 9, 357–359 (2012). doi:10.1038/nmeth.1923 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
80 吴浩 等,从无重复的全基因组亚硫酸氢钠测序数据中检测差异甲基化区域。 核酸研究 43,e141(2015)。 doi:10.1093/nar/gkv715 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
81. 拉米雷斯F 等,deepTools2:用于深度排序数据分析的下一代web服务器。 核酸研究 44,W160–W165(2016)。 doi:10.1093/nar/gkw257 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
82 Buenrostro JD,Giresi PG,Zaba LC,Chang HY,Greenleaf WJ,天然染色质的转位,用于开放染色质、DNA结合蛋白和核小体位置的快速灵敏表观基因组分析。 自然方法 10, 1213–1218 (2013). doi:10.1038/nmeth.2688 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
83 加伯M 等,通过利用偏置替代模式识别新的约束元素。 生物信息学 25,i54–i62(2009年)。 doi:10.1093/bioinformatics/btp190 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
84 米·H 等人,PANTHER版本11:来自基因本体论和反应体途径的扩展注释数据,以及数据分析工具的增强。 核酸研究 45,D183–D189(2017)。 doi:10.1093/nar/gkw1138 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
85 萨维茨基P 等人,《结晶用人类蛋白质的高通量生产:SGC的经验》。 J.结构。 生物 172, 3–13 (2010). doi:10.1016/j.jsb.2010.06.008 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
86 Bourenkov GP,Popov AN,《数据收集策略的定量方法》。 《水晶学报》。 D类 62, 58–64 (2006). doi:10.1010/S0907444905033998 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
87 卡布施W,XDS。 《水晶学报》。 D类 66, 125–132 (2010). 网址:10.1107/S0907444909047337 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
88 Winn医学博士 等人,CCP4套件概述和当前发展。 《水晶学报》。 D类 67, 235–242 (2011). doi:10.1107/S0907444910045749 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
89 麦考伊AJ 等人,Phaser晶体学软件。 J.应用。 Crystalogr公司 40, 658–674 (2007). doi:10.1107/S0021889807021206 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
90 亚当斯PD 等,PHENIX:基于Python的大分子结构溶液综合系统。 《水晶学报》。 D类 66, 213–221 (2010). doi:10.1107/S0907444909052925 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
91 Emsley P、Cowtan K、Coot:分子图形的建模工具。 《水晶学报》。 D类 60, 2126–2132 (2004). doi:10.1107/S0907444904019158 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
92 Emsley P、Lohkamp B、Scott WG、Cowtan K,《Coot的特点和发展》。 《水晶学报》。 D类 66, 486–501 (2010). doi:10.1107/S0907444910007493 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
93 Afonine光伏 等人,《使用菲的自动晶体结构精细化》。 《水晶学报》。 D类 68, 352–367 (2012). doi:10.1107/S090744912001308 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
94 Vagin AA,Isupov MN,球平均相移函数及其在电子密度图中分子和碎片搜索中的应用。 《水晶学报》。 D类 57, 1451–1456 (2001). doi:10.1107/S090744490102409 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
关联数据 本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。