介绍 FET家族基因 保险丝 , EWSR1、, 和 TAF15型 (也称为 TLS公司 , EWS中, 和 TAF2N公司 分别)编码RNA结合蛋白(图 1 A) 被提议将转录与RNA剪接、加工和转运的后续步骤联系起来 1 , 2 , 三 , 4 , 5 ,本地化翻译 6 和微RNA处理 7 将FET基因作为5′伙伴和替代转录因子编码基因作为3′伙伴的融合癌基因(图 1 B) 是多种类型肉瘤和白血病的病理学特征 8 , 9 FET致癌基因引起的肿瘤几乎没有其他突变,这表明FET融合癌蛋白对肿瘤发展的关键机制有影响 9 , 10 , 11 , 12 , 13 , 14 , 15 .
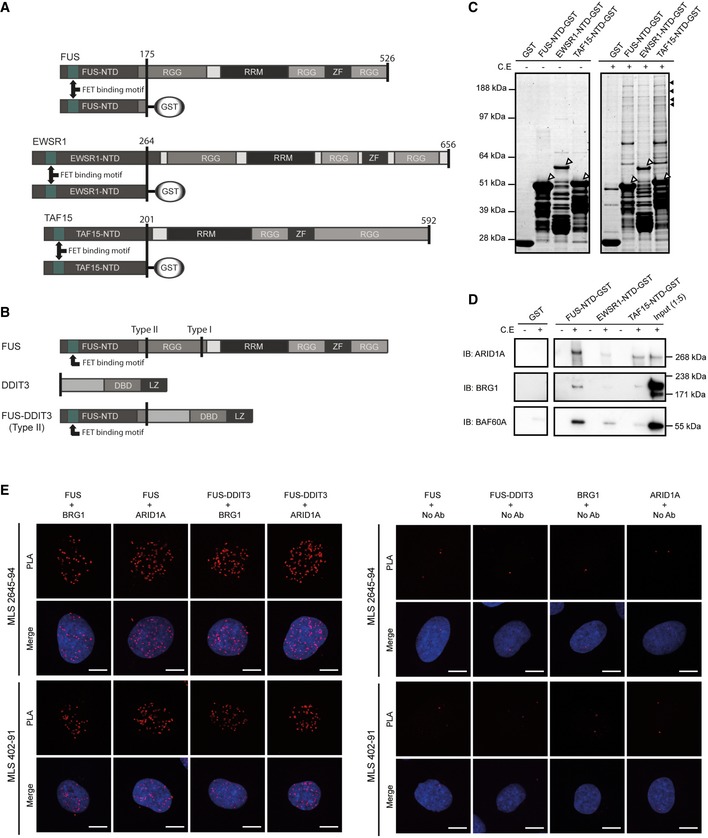
图1。 识别与重组FET‐NTD毒饵结合的蛋白质。
这三种正常的RNA结合FET蛋白由N端重复和结构无序域(NTD:N端域)、中心RNA结合域(RRM:RNA识别基序)、锌指域(ZF)和退化重复区(RGG:RGG重复区)组成。 显示的GST标记FET‐NTD毒饵用于下拉实验,代表FET融合癌蛋白中常见的最短部分。 FET结合基序是三种FET蛋白之间形成复合物所需的保守序列。 显示氨基酸数量。
典型FET融合蛋白(FUS‐DDIT3 II型)及其亲本蛋白的示意图。 DBD:DNA结合域,LZ:亮氨酸拉链域。 I型和II型显示了FUS中两个最常见的MLS融合断点的位置。
使用GST标记的FET‐NTD作为诱饵,对SDS–PAGE分离的下拉样品进行考马斯染色,包括/不包括细胞提取物(C.E)。 含有结合GST的Sepharose作为对照。 左侧面板(C.E-)显示了重组蛋白毒饵(用白色箭头表示)以及较小部分重组产品的背景。 FET‐NTD(C.E+)保留了几个高分子量蛋白质。 黑色箭头表示通过质谱法分析的蛋白质带和凝胶部分(另请参见 表EV1 ). 使用FET‐NTD诱饵和/或不使用细胞提取物(C.E)的下拉样品的免疫印迹分析(IB)。 使用抗ARID1A、BRG1和BAF60A的抗体检测SWI/SNF成分。 以1:5稀释的输入样品作为对照。
现场 使用抗BRG1、ARID1A、DDIT3和正常FUS C端部分的抗体进行的邻近连接分析(PLA)显示,含有FUS/BRG1、FUS/ARID1A、FUS-DDIT3/BRG1和FUS-DID3/ARID1A的蛋白质复合物是MLS细胞系2645-94和402-91细胞核中的红色荧光点。 FUS‐DDIT3融合蛋白中不存在FUS的C端部分,正常的DDIT3在这些细胞系中不表达。 合并后的图像还包括蓝色DAPI核复染。 主要抗体组合用于检测相互作用(左侧面板)。 在对照实验(右侧面板)中,省略一个一级抗体来评估背景荧光信号。 比例尺=10μm。
可在线获取此图的源数据。
FET融合癌蛋白总是包含FET伴侣的N末端结构域(NTD),与转录因子伴侣的DNA结合部分并列(图 1 A和B)。 据报道,它们作为异常转录因子发挥作用,NTD作为强转录激活域 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 FET癌基因的强制表达或沉默影响肿瘤形态和大量基因的调控,并改变表观遗传景观 24 , 25 , 26 , 27 .
三种正常的FET蛋白包含中央RNA识别基序(RRM),其两侧是RGG重复区和潜在的单链DNA或RNA结合锌指结构域(图 1 A) 28 , 29 , 30 NTD主要由结构无序、类朊蛋白退化的富含SYGQ的重复序列组成,其组成表明其在蛋白质相互作用中具有功能 31 在一些FET融合癌蛋白和肿瘤实体中,它们作为N末端伴侣在功能上相互取代,这进一步突显了它们的相似性 28 , 32 根据这些观察结果,我们假设三个FET‐NTD可以通过绑定相同的关键交互伙伴来发挥作用。 然而,即使发现了几个FET结合蛋白,也没有报道过这种相互作用伙伴,NTD的作用仍然是个谜。 本研究的目的是确定三种FET‐NTD共享的主要相互作用伙伴,这些伙伴可能为共同的发病机制提供线索。
结果和讨论 FET‐NTD介导正常和致癌FET蛋白与SWI/SNF的结合 我们在细胞提取物的下拉实验中使用细菌表达的GST标记的重组构建物,对FET‐NTD结合蛋白的富集进行无偏见的分析(图 1 A) ●●●●。 SDS–PAGE分析和蛋白质染色显示,三种FET‐NTD都捕获了几个高分子量蛋白质(图 1 C) ●●●●。 切割凝胶带的质谱分析(MS)确定了SWI/SNF染色质重塑复合物核心成分的肽:ARID1A(BAF250A)、BRG1(SMARCA4)、BAF170(SMARCC2)和BAF155(SMARCC)( 表EV1 ). 免疫印迹分析也证实SWI/SNF核心成分的富集(图 1 D) ●●●●。 综上所述,结果表明所有三个FET‐NTD都有能力结合SWI/SNF染色质重塑复合物。 这种大型多亚单位复合物由大约15个紧密结合的核心蛋白组成,这些核心蛋白通过ATP驱动的核小体重新定位和抑制多梳复合物的清除来控制基因表达 33 , 34 从密切相关的基因中引入替代核心蛋白会产生许多不同的SWI/SNF复合物。 SWI/SNF活性在细胞和组织中的重要性从敲除实验和影响多种癌症中SWI/SNF-成分的突变中可以明显看出 35 .
正常FET蛋白在细胞核和细胞质之间穿梭,而融合癌蛋白主要局限于细胞核 5 , 36 , 37 . 现场 近接结扎分析显示黏液样脂肪肉瘤(MLS)肿瘤细胞中含有与SWI/SNF核心蛋白BRG1和ARID1A结合的正常FUS或致癌FUS‐DDIT3的核复合体(图 1 E) 。 结果证实了正常FUS和FUS‐DDIT3癌蛋白与SWI/SNF的结合 就地 并显示这些复合物的核定位。
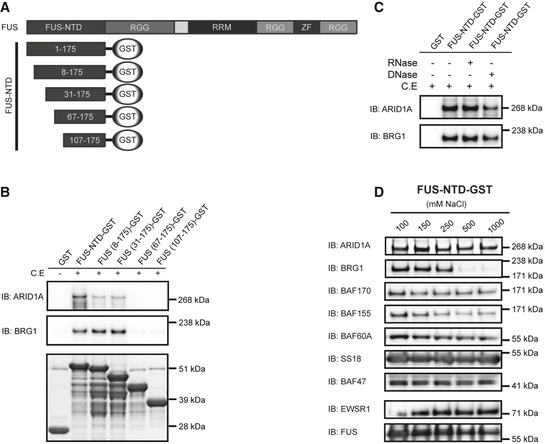
与SWI/SNF的相互作用强大,并由FUS‐NTD中的保守基序介导 为了进一步分析FET‐NTD结合特性,我们选择了FUS‐NTD,因为它是最短的致癌NTD变体,与EWSR1和TAF15 NTD的主要区别在于退化的富含SYGQ的重复序列的数量 28 , 31 , 38 为了定义结合SWI/SNF的FET‐NTD序列,我们构建了一系列FUS‐NTT缺失突变毒饵(图 2 A) ●●●●。 当氨基酸31–66被删除时,与SWI/SNF核心蛋白的结合失败(图 2 B) ●●●●。 该序列包含一个保守的26个氨基酸的“FET结合基序”,对三种正常FET蛋白和细胞质蛋白凝集素之间形成同源或异质复合物也很重要(图 1 A) 31 , 39 DNase和RNase处理均未中断FUS‐NTD与SWI/SNF的结合,不包括作为相互作用介质的核酸(图 2 C) ●●●●。 综上所述,这些结果表明FET蛋白与SWI/SNF的结合独立于核酸,并且只有一小部分重复结构域可能足以进行结合。 结果与结合SWI/SNF的FET蛋白兼容,无论是作为单分子,还是作为包括融合癌蛋白的多聚FET复合物。
图2。 FUS‐NTD和SWI/SNF核心成分之间的结合分析。
FUS‐NTD‐GST截断构造的示意图。 显示氨基酸数量。
GST下拉样品的免疫印迹分析(IB),将FUS‐NTD‐GST缺失突变体作为含有/不含细胞提取物(C.E)的诱饵。 ARID1A和BRG1被用作SWI/SNF结合的示踪剂。 重组蛋白的考马斯染色如下图所示。 显示氨基酸数量。
使用抗ARID1A和BRG1抗体,对RNase或DNase处理后的FUS‐NTD‐GST‐细胞提取物(C.E.)下拉样品进行免疫印迹分析(IB)。
增加100、150、250、500或1000 mM NaCl的严格洗涤后,SWI/SNF组分(ARID1A、BRG1、BAF170、BAF155、BAF60A、SS18和BAF47)和全长FET蛋白(EWSR1和FUS)在sepharose结合的FUS‐NTD‐GST‐下拉毒饵上的免疫印迹分析(IB)。 注意NaCl浓度>250 mM时BRG1的损失。
可在线获取此图的源数据。
通过增加NaCl浓度的严格清洗,进一步测试SWI/SNF与重组FUS‐NTD的结合。 洗脱液的免疫印迹分析显示,在250 mM NaCl以上时BRG1组分丢失,而ARID1A、BAF170、BAF155、BAF60A、BAF47、SS18、FUS和EWSR1在1 M盐下仍与重组FUS‐NTD结合(图 2 D) ●●●●。 这表明FET‐NTD与几个SWI/SNF核心蛋白的结合非常牢固,但BRG1通过其他SWI/SNF组分的结合较弱,可能是间接结合。
FET癌蛋白的主要组分与SWI/SNF相互作用,与正常FET蛋白无结合竞争 为了进一步证实正常和致癌FET蛋白的SWI/SNF结合,我们使用BRG1特异性抗体对携带四种不同FET癌基因的MLS和尤因肉瘤(EWS)细胞系的SWI/SNF复合物进行免疫沉淀(IP)( 保险丝‐DDIT3 类型I, 保险丝‐DDIT3 II型, EWSR1‐FLI1, 和 EWSR1‐ERG公司 )(表 1 ,图 电动汽车1 A和B以及 表EV2 ). 免疫沉淀物的MS和免疫印迹分析显示存在正常FUS和EWSR1,以及致癌FUS‐DDIT3和EWSR‐FLI1/ERG,证实正常和致癌FET蛋白结合SWI/SNF复合物。 分析还表明,这些肿瘤细胞株产生了整套SWI/SNF核心蛋白(表 1 和 表EV2 ).
表1。 经质谱(MS)和关键蛋白免疫印迹(IB)验证的MLS 402‐91、MLS 2645‐94、EWS TC‐71和EWS IOR/CAR的DDIT3 Co‐IP后和EWS TC71的FLI1 Co‐的结合部分中SWI/SNF成分和FET蛋白(野生型和融合癌蛋白)的总结。 请注意,一些SWI/SNF组件由几个备选变体之一表示,例如BAF45A‐D。 每个质谱点击的肽列表如所示 表EV2 和 电动汽车3
蛋白质 备选名称 BRG1公司IP DDIT3合作IP FLI1合作IP
英里数402‐91 MLS 2645‐94号 EWS TC‐71 EWS IOR/CAR MLS 402‐91号 EWS TC‐71
BRG1公司 SMARCA4系统 理学硕士、文学学士 理学硕士、文学学士 理学硕士、文学学士 毫秒,IB 理学硕士、文学学士 理学硕士、文学学士
BRM公司 SMARCA2系列 ‐ ‐ ‐ ‐ (微软) 微软
ARID1A公司 BAF250A型 微软 微软 微软 微软 微软 微软
ARID1B公司 BAF250B型 ‐ 微软 ‐ ‐ 微软 微软
ARID2公司 BAF200型 ‐ ‐ 微软 微软 微软 微软
BAF170型 SMARCC2系统 微软 微软 微软 微软 微软 微软
BAF155型 SMARCC1型 微软 微软 微软 微软 微软 微软
BAF60A型 SMARCD1系统 微软 微软 微软 微软 微软 微软
BAF60B型 SMARCD2系统 微软 微软 微软 微软 微软 微软
BAF60C型 SMARCD3系统 微软 ‐ 微软 微软 (微软) 微软
BAF57型 智能电网1 微软 微软 微软 微软 微软 微软
BAF47型 SMARCB1型 微软 微软 微软 微软 微软 理学硕士、文学学士
BAF53A公司 ACTL6A系列 微软 微软 微软 微软 微软 微软
BAF53B公司 ACTL6B系列 ‐ ‐ ‐ ‐ ‐ ‐
BAF45A型 10菲律宾比索 ‐ ‐ 微软 微软 ‐ 微软
BAF45B型 DPF1(DPF1) ‐ ‐ ‐ ‐ ‐ ‐
BAF45C型 DPF3(DPF3) ‐ ‐ ‐ ‐ 微软 ‐
baf45天 DPF2型 微软 微软 微软 微软 微软 微软
BAF180型 PBRM1项目 微软 微软 微软 微软 微软 微软
2018春夏 SYT公司 ‐、IB 微软 微软 微软 微软 ‐、IB
保险丝 TLS公司 微软 微软 微软 微软 微软 微软
EWSR1号机组 预警系统 理学硕士、文学学士 毫秒,IB 理学硕士、文学学士 理学硕士、文学学士 微软 微软
TAF15型 TAF2N公司 ‐ 微软 微软 微软 微软 微软
保险丝‐DDIT3 TLS‐CHOP公司 ‐、IB ‐、IB 理学硕士、文学学士
EWSR1‐FLI1号机组 ‐、IB 理学硕士、文学学士
EWSR1‐ERG公司 微软
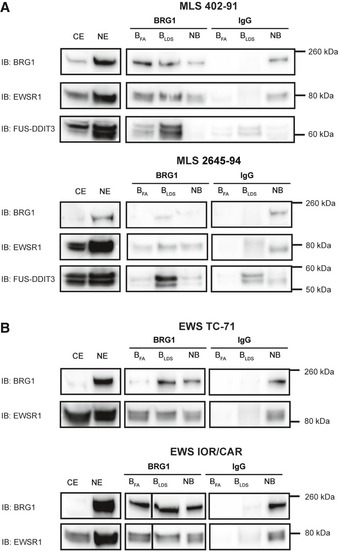
图EV1。 肉瘤细胞系的BRG1 Co‐IP用甲酸洗脱。
与BRG1共免疫沉淀蛋白的免疫印迹分析(IB)。 在MLS细胞系402‐91和2645‐94中检测BRG1、EWSR1和FUS‐DDIT3抗体。
与BRG1共免疫沉淀蛋白的免疫印迹分析(IB)。 用EWS细胞株TC‐71和IOR/CAR中的BRG1和EWSR1抗体进行检测。 CE:细胞质提取物,NE:核提取物,B FA公司 :用1%甲酸洗脱结合蛋白,B LDS公司 :用LDS样品缓冲液洗脱的结合蛋白,NB:非结合蛋白,未被抗体捕获。
可在线获取此图的源数据。
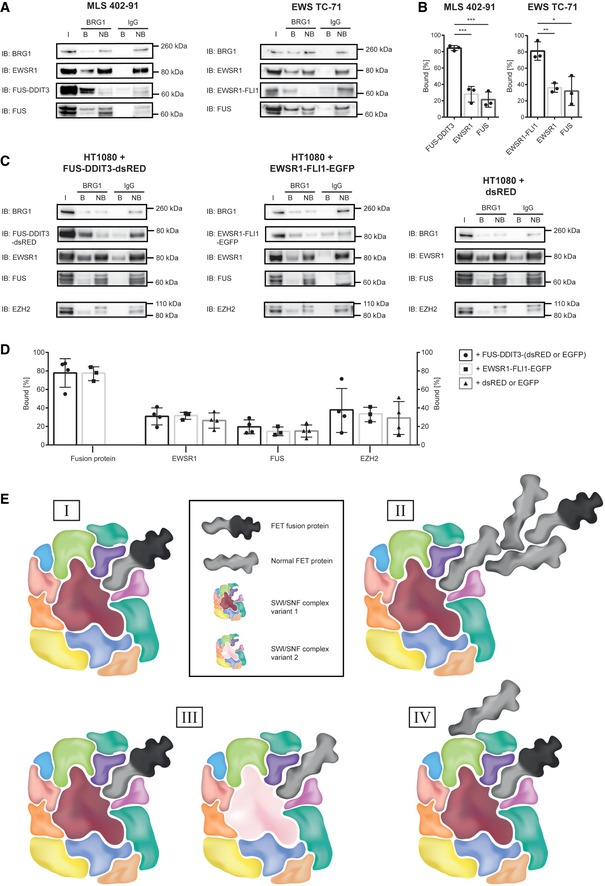
然后,我们使用抗BRG1 IP来量化表达FUS‐DDIT3和EWSR1‐FLI1的细胞系中FET蛋白和SWI/SNF之间的相互作用(图 三 A和B)。 正常FET蛋白和SWI/SNF复合物在大多数组织和细胞类型中丰富,根据我们的结果,FET‐NTD与SWI/SNF结合,它们之间可能会发生广泛的相互作用。 然而,只有少数正常FET蛋白与沉淀的SWI/SNF结合,这表明正常FET蛋白质结合受到调控,可能局限于染色质位置、活性或SWI/SNF-复合物亚型。 相反,FET癌蛋白表达较弱 40 但主要组分与BRG1沉淀复合物结合,表明与SWI/SNF的相互作用失调。 此外,温和的甲酸洗脱释放出大量EWSR1,而用强LDS洗涤剂洗脱需要从抗BRG1捕获的复合物中释放FUS‐DDIT3融合蛋白(图 电动汽车1 A和B)。 综上所述,这些结果表明,与正常FET蛋白相比,更多的FET癌蛋白与SWI/SNF结合,结合强度增加。
图3。 肉瘤细胞系中SWI/SNF和FET蛋白的免疫共沉淀。
与MLS细胞系402‐91和EWS细胞系TC‐71核提取SWI/SNF共免疫沉淀蛋白的免疫印迹分析(IB)。 使用抗BRG1、DDIT3(FUS‐DDIT3)、正常FUS和EWSR1的C端部分以及FLI1(EWSR1‐FLI1)的抗体进行检测。 为了直接量化结合蛋白和非结合蛋白的比例,将每个IP样品的相对蛋白质量加载到凝胶上,并考虑免疫沉淀过程中的稀释。 I: 核提取物的输入,B:结合的蛋白质,NB:未结合的蛋白质。 显示了一个具有代表性的免疫印迹。 源数据中显示了所有复制品的免疫印迹。
图中显示了MLS 402‐91细胞系中FUS‐DDIT3、EWSR1和FUS以及EWS TC‐71细胞系中EWSR1‐FLI1、EWSRl和FUS的免疫印迹结合信号强度占总信号强度的百分比。 平均值±SEM用圆圈表示单个重复( n个 = 3). 学生的 t吨 ‐测试* P(P) < 0.05, ** P(P) < 0.01, *** P(P) < 0.001. 所有量化的原始数据,包括 P(P) 值显示为源数据。
与核提取SWI/SNF共免疫沉淀的蛋白质的免疫印迹分析显示为具有代表性的免疫印记,这些蛋白质来自具有过度表达FUS‐DDIT3‐dsRED、EWSR1‐FLI1‐EGFP或dsRED的模型细胞系HT1080。 使用抗BRG1、DDIT3(FUS‐DDIT3‐dsRED)、正常FUS的C端部分以及EWSR1、EZH2和FLI1(EWSR1‐FLI1‐EGFP)的抗体进行检测。 为了直接量化结合蛋白和非结合蛋白的比例,将每个IP样品的相对蛋白质量加载到凝胶上,并考虑免疫沉淀过程中的稀释。 一: 输入核提取物,B:结合蛋白,NB:未结合蛋白。 源数据中显示了所有复制品的免疫印迹(表达了dsRED或EGFP标记的蛋白质)。
图中显示了免疫印迹中融合蛋白(FUS‐DDIT3或EWSR1‐FLI1)、EWSR1、FUS和EZH2的结合信号强度占总信号强度(结合信号强度+非结合信号强度)的百分比,该融合蛋白在过度表达FUS-DDIT3‐dsRED/‐EGFP的模型细胞系HT1080中( n个 =4,圆圈),EWSR1‐FLI1‐EGFP( n个 =3,正方形)或dsRED/EGFP( n个 =4,三角形)。 显示平均值±SEM,并显示单个重复。 学生的 t吨 ‐测试,无重大变化, P(P) > 0.05. 所有量化的原始数据,包括 P(P) 值显示为源数据。
正常和致癌FET蛋白与SWI/SNF结合的四种替代模型。 (一) :FET癌蛋白直接与SWI/SNF结合。 (二) :正常FET蛋白形成同源和异源复合物,并介导FET癌蛋白的结合。 (三) :FET癌蛋白和正常FET蛋白结合到具有不同生化成分的SWI/SNF复合物的不同变体。 (IV) :FET癌蛋白和正常FET蛋白与SWI/SNF复合物上的不同位点结合。
可在线获取此图的源数据。
正常和致癌FET蛋白与SWI/SNF复合物的结合可能导致对结合位点的竞争。 为了验证这一假设,我们在人类HT1080纤维肉瘤细胞中表达了DsRED或EGFP标记的FUS‐DDIT3、EWSR1‐FLI1或单独的标记,并量化了与BRG1共沉淀的正常和致癌FET蛋白。 虽然结果显示FET癌蛋白与BRG1沉淀物大量结合,但未观察到BRG1结合的FUS或EWSR1蛋白的减少(图 三 C和D)。
基于我们的综合结果,我们提出了FET融合癌蛋白与SWI/SNF染色质重塑复合物之间结合的替代模型(图 三 E) 。 正常和致癌FET蛋白之间不同的IP结合/洗脱模式表明,融合癌蛋白直接与SWI/SNF结合,而不是通过与正常FET蛋白的多聚间接结合(图 三 E、 型号I和II)。 缺乏竞争也表明正常和致癌FET蛋白结合SWI/SNF的不同变体或结合SWI/SNF复合物上的不同位点(图 三 E、 模型III和IV)。 如前所述,FET癌蛋白直接与正常FET蛋白结合 31 和正常FET蛋白形成同源和异源复合物。 此外,SWI/SNF复合物的许多共存变体 35 , 41 可能具有不同的绑定属性。 因此,FET蛋白和SWI/SNF变体之间的几种替代复合物可能在肿瘤细胞中共存。
FET癌蛋白结合的SWI/SNF复合物显示出正常的核心蛋白组成 滑膜肉瘤中的SS18-SSX融合癌蛋白改变了SWI/SNF复合物的组成。 含有SS18 SWI/SNF核心蛋白N端部分的融合蛋白将BAF47蛋白从复合物中清除 42 , 43 。即使FET蛋白或其融合伙伴不被视为SWI/SNF核心成分 35 理论上,它们可能导致SWI/SNF组成发生变化。 为了测试这种可能性,我们使用DDIT3和FLI1特异性抗体从MLS和EWS细胞系中沉淀FUS‐DDIT3与EWSR1‐FLI1癌蛋白。 由于这些细胞系中没有正常的DDIT3或FLI1表达,抗体只沉淀融合癌蛋白及其相互作用蛋白。 沉淀物的MS分析表明,SWI/SNF核心蛋白与融合癌蛋白共沉淀而成(表 1 和 表EV3 ). 这些结果排除了致癌FET蛋白导致SWI/SNF核心蛋白丢失的可能性,并表明了与SS18-SSX融合癌蛋白不同的作用机制 42 , 43 它还表明,FET癌蛋白与几个SWI/SNF变体结合,因为在细胞中发现的所有替代SWI/SNF-核心蛋白也与FET癌蛋白质沉淀。
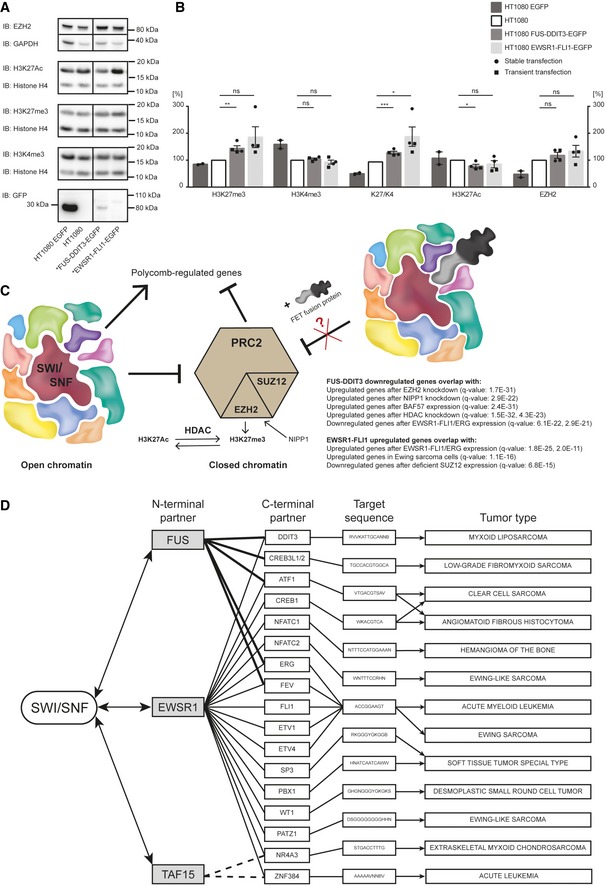
FET癌蛋白的强制表达导致H3K27me3水平升高 FET癌蛋白与SWI/SNF的结合促使我们研究这是否会导致任何功能性影响。 据报道,SWI/SNF可以平衡PRC2多梳阻遏物复合物引起的抑制性H3K27三甲基化(H3K27me3) 33 , 34 据报道,SWI/SNF成分的致癌突变会破坏这种平衡,导致多梳活性和H3K27me3水平增加 34 例如,在缺乏SWI/SNF核心BAF47/SMARCB1蛋白的细胞中报告H3K27me3升高 44 我们对人类HT1080肉瘤细胞蛋白提取物中组蛋白修饰的免疫印迹分析表明,癌基因的强制表达 保险丝‐DDIT3 导致H3K27me3水平显著升高(图 4 A和B,以及图中的完整数据集 电动汽车2 A–I)。 EWSR1‐FLI1号机组 转染也导致H3K27me3水平增加但更可变。 其他实验验证了融合癌蛋白诱导的H3K27三甲基化(图 电动汽车2 C、 D、H和I)。 用EZH2抑制剂tazemetostat处理细胞后,H3K27me3水平显著降低,证明定量Western blot分析是有效的(图 电动汽车2 E和J)。 H3K4me3水平基本上不受影响,对H3K27乙酰化的影响有所不同,但没有一致的趋势。 我们观察到EZH2表达略有增加,但无统计学意义(图 4 A和B),而与BRG1的共沉淀没有增加(图 三 C和D)。 对这些结果的合理解释是,FET癌蛋白与SWI/SNF的结合对其PRC2平衡功能产生负面影响,导致PRC2/EZH2活性增加。 因此,观察到的FET癌基因效应与SWI/SNF核心复合蛋白中某些致癌突变引起的效应相似。 H3K27me3水平的增加可能看起来微不足道,但可能转化为数百个基因调控的改变。
图4。 FET致癌基因诱导的H3K27三甲基化变化和下游效应。
稳定转染表达EGFP、FUS-DDIT3-EGFP或EWSR1-FLI1-EGFP的HT1080细胞系组蛋白修饰的免疫印迹分析(IB)。 针对H3K27Ac、H3K17me3、H3K 4me3和组蛋白负载控制H4的抗体用于检测组蛋白修饰和针对EZH2的抗体,负载控制GAPDH用于评估催化PRC2量。 用GFP抗体进行免疫印迹分析可以验证FET融合癌蛋白和EGFP的表达。 显示了一个具有代表性的免疫印迹。 图中显示了更多的免疫印迹,包括第二个稳定的生物复制和瞬时转染(24和48小时)后组蛋白修饰的分析 电动汽车2 A–D。 显示根据免疫印迹相对每个相应的负载控制H4或GAPDH量化的蛋白质量(H3K27me3、H3K4me3、比率H3K17me3/H3K4me3、H3G27Ac和EZH2)的图表,归一化为亲本HT1080。 平均值±扫描电镜显示了稳定转染的两个实验中的单个重复(见图 4 A、 和 电动汽车2 A和F)和两个瞬时转染实验(24和48小时,见图 电动汽车2 B和G), n个 = 4. 学生的 t吨 ‐测试,ns=不显著* P(P) < 0.05, ** P(P) < 0.01, *** P(P) <0.001。 所有量化的原始数据,包括 P(P) 值显示为源数据。 SWI/SNF反对多梳复合体PRC2。 PRC2的催化亚单位EZH2催化组蛋白H3(H3K27me3)上Lys27的三甲基化,这是一种与封闭染色质相关的染色质修饰,导致多梳调控基因的下调。 FET与SWI/SNF的融合结合可能会损害SWI/SNF的功能,包括多梳对抗。 特别是,当FUS‐DDIT3在HT1080细胞中过度表达时,基因下调(RNA‐seq数据, n个 =3,与对照组相比, n个 =4)EZH2敲除、NIPP1敲除和HDAC敲除以及SMARCE1空细胞系中BAF57(SMARCEl)重建后与上调基因集重叠。 HT1080 wt的RNA-seq数据( n个 =4),HT1080 EGFP( n个 =4),HT1080 FUS‐ddid3‐EGFP( n个 =3)和HT1080 EWSR1‐FLI1‐EGFP( n个 =4),并使用分子签名数据库(MSigDB)分析>双重调控的基因。 基因列表与基因集集合“化学和遗传扰动”进行了比较,源数据中显示了每次比较的前20个基因集。 q个 值(FDR调整 P(P) ‐值)在括号中表示。
融合癌蛋白FET家族、靶向DNA序列和相关肿瘤的示意图。 左图:FET N端融合伴侣并与SWI/SNF结合。 中心:C末端转录因子融合伙伴及其DNA靶序列(JASPAR数据库)。 右:由各自融合癌基因引起的肿瘤类型。 注意每个融合癌蛋白的肿瘤类型特异性,FET‐NTD在某些实体中作为融合伙伴相互替换。 随着越来越多的成员被不断发现,只有一部分FET家族融合癌蛋白和肿瘤被显示出来。
可在线获取此图的源数据。
图EV2。 组蛋白H3修饰分析。
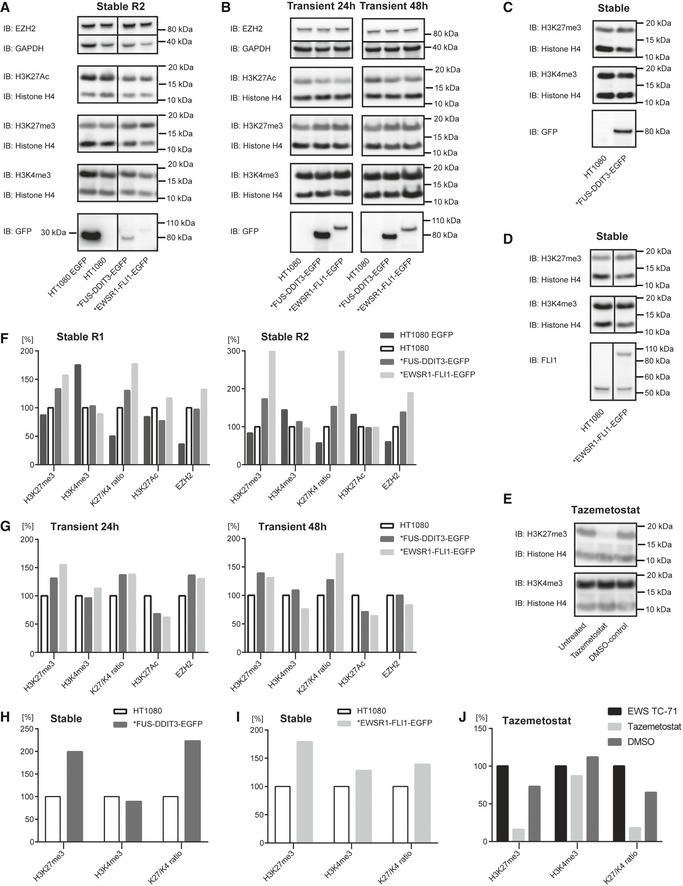
A类 稳定转染表达EGFP、FUS‐DDIT3‐EGFP或EWSR1‐FLI1‐EGFP的HT1080细胞系复制R2组蛋白修饰的免疫印迹分析(IB)。 针对H3K27Ac、H3K17me3、H3K 4me3和组蛋白负载控制H4的抗体用于检测组蛋白修饰和针对EZH2的抗体,负载控制GAPDH用于评估催化PRC2量。 用GFP抗体进行免疫印迹分析可以验证FET融合癌蛋白和EGFP的表达。 稳定的重复R1如图所示 4 答:。
B类 HT1080和HT1080瞬时表达FUS‐DDIT3‐EGFP或EWSR1‐FLI1‐EGFP(瞬时转染24或48小时后采集的样品)的免疫印迹分析(IB),以及抗H3K27Ac、H3K27 me3、H3K 4me3抗体, 组蛋白负载控制H4用于检测组蛋白修饰和EZH2抗体,负载控制GAPDH用于评估催化PRC2量。 用GFP抗体进行免疫印迹分析可以验证FET融合蛋白的表达。
C、 D类 HT1080和HT1080 FUS‐DDIT3‐EGFP(C)或HT1080 EWSR1‐FLI1‐EGFP(D)的免疫印迹分析(IB)(均为稳定表达),带有针对H3K27me3、H3K4me3和组蛋白负载控制H4的抗体。 用GFP或FLI1抗体进行免疫印迹分析,以验证FET融合蛋白和正常FLI1的表达。 注意,内源性FLI1约50 kDa在HT1080细胞系中表达。
E类 未经处理的EWS TC‐71提取物、用5μM他汀类药物处理72小时的细胞和DMSO对照的免疫印迹分析(IB)。 使用针对H3K27me3、H3K4me3和组蛋白负载控制H4的抗体进行检测,结果表明,在使用他汀类药物抑制EZH2后,H3K17me3显著降低。
F类 显示数量的图表,从图中的免疫印迹定量 4 与HT1080 EGFP、HT1080 FUS‐DDIT3‐EGFP和HT1080 EWSR1‐FLI1‐EGFF的负载控制GAPDH相比,H3K27me3、H3K4me3和H3K27 Ac(与相应的组蛋白负载控制H4相关)的A(R1)或面板(A)(R2)、H3K 27me3/H3K4me3以及EZH2的数量。 每个图表面板显示一个实验的数据。
G公司 图表显示了H3K27me3、H3K4me3和H3K17Ac(与相应的组蛋白负载控制H4相关)的数量,H3K24me3/H3K4me3的比率,以及与HT1080 FUS‐DDIT3‐EGFP和HT1080 EWSR1‐FLI1‐EGFP的负载控制GAPDH相比EZH2的数量,这些数据来自于面板(B)中的免疫印迹。 在24小时或48小时瞬时转染后采集样本。 每个图表面板显示一个实验的数据。
H、 我 显示H3K27me3和H3K4me3的量(相对于相应的组蛋白负载对照H4)以及从面板(C)量化的HT1080 FUS‐DDIT3‐EGFP(H)或从面板(D)量化的HT1080 EWSR1‐FLI1‐EGFP(I)的H3K27me3/H3K4me3比率的图(均为稳定表达)标准化为亲本HT1080。 每个图表面板显示一个实验的数据。
J型 图中显示了H3K27me3和H3K4me3(与相应的组蛋白负载量控制H4相关)的数量,以及未经治疗的EWS TC‐71、用5μM他汀治疗72小时的细胞和DMSO控制的H3K17me3/H3K4me3比率,并将其归一化为未经治疗对照。 来自一个实验的数据。
可在线获取此图的源数据。
我们的结果与最近发表的关于EWS细胞系的研究结果部分不一致,报告称对H3K27me3水平没有影响,而是增加了特定基因组位点的H3K27乙酰化水平 24 , 45 分歧结果可以通过使用不同的实验系统来解释。 正常FLI1蛋白在我们研究中使用的HT1080细胞中组成性表达(图 电动汽车2 D) 而EWS细胞中没有这种蛋白 46 。组成性FLI1表达可阻断EWSR1‐FLI1结合和该癌蛋白诱导的组蛋白乙酰化作用。 此外,用于EWS特定研究的基于ChIP‐seq的分析显示了基因组位置和组蛋白修饰的分布,但可能无法检测到H3K27me3中微小但广泛的变化。
FUS‐DDIT3改变的基因表达模式与PRC2调节的基因集重叠 进一步研究FET致癌基因的下游效应 保险丝‐DDIT3 和 EWSR1‐FLI1, 我们通过RNA‐seq来比较FET癌基因转染和野生型HT1080细胞的基因表达模式( n个 =3–4)(图 4 C和源数据)。 使用基因集富集分析(GSEA)工具,将>2倍调控基因列表与“化学和遗传扰动”(CGP,3433个基因集)后改变的基因集数据库进行比较。 敲除PRC2组分EZH2后,FUS‐DDIT3下调基因与细胞内上调基因集显著重叠( q个 ‐值1.68×10 −31 )或重组SWI/SNF组分BAF57/SMARCE1( q个 ‐值2.37×10 −31 ). NIPP1(蛋白磷酸酶核抑制剂-1)的缺失导致EZH2表达的降解 47 与这一观察结果一致,FUS‐DDIT3下调的基因与NIPP1敲低后上调的基因显著重叠( q个 值2.9×10 −22 ),进一步强调了对PRC2活性的影响。 这些结果与我们的假设一致,即FUS‐DDIT3诱导的基因下调涉及SWI/SNF‐PRC2平衡的破坏(图 4 C) ●●●●。 相反,FUS‐DDIT3上调基因与任何显著性相对较低的基因集没有重叠。
EWSR1‐FLI1调控基因与EWSR1−FLIl表达被修饰的条件下的基因集存在显著重叠。 此外,我们上调的基因与包含EWSR1‐FLI1上调基因集的CGP基因集重叠,我们下调的基因与含有EWSR1−FLI下调基因的CPG集匹配,这表明EWSR1∙FLI转染HT1080细胞模拟EWS细胞(图 4 C和源数据)。 与表达FUS‐DDIT3的细胞相比 q个 EWSR1‐FLI调节基因列表和PRC2‐或SWI/SNF调节条件下的基因集之间的得分重叠。 这表明FUS‐DDIT3和EWSR1‐FLI1在HT1080细胞中的作用不同,尽管SWI/SNF是这两种癌蛋白的主要结合伙伴。
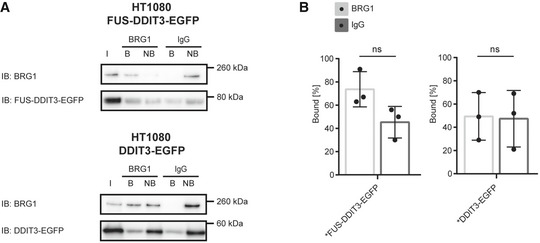
FUS‐DDIT3和EWSR1‐FLI1之间的差异效应可以用DDIT3与FLI1转录因子伙伴的非常不同的性质来解释。 DDIT3是CEBP家族的一种含亮氨酸拉链的二聚体形成转录因子。 然而,它有一个酸性的DNA结合域,因此被认为发挥主要的负功能,阻止大多数二聚体伴侣的DNA结合 48 支持这一观点,DDIT3的异位表达主要导致受影响基因的负调控 25 , 49 然而,内质网应激诱导DDIT3和二聚体伴侣ATF4的强共表达,从而产生DNA结合二聚体对 50 在MLS中,DDIT3在没有ATF4的情况下在体外表达,DNA靶位点的数量有限。 相反,EWSR1‐FII1可以结合基因组中成千上万个潜在的DNA结合位点 51 , 52 ,并且由此产生的SWI/SNF募集和激活这些位点的增强子被认为是致癌机制的重要部分。 据报道,正常FLI1蛋白与SWI/SNF自身结合,从而有助于FET癌蛋白结合。 在这里,我们测试了正常的DDIT3是否能结合SWI/SNF。 在稳定表达GFP标记的DDIT3的HT1080细胞中的实验表明,DDIT3与BRG1的共沉淀较弱(图 电动汽车3 A和B)。 这些结果表明,FUS和DDIT3部分都有助于FUS‐DDIT3与SWI/SNF的强结合。 因此,FUS‐DDIT3和EWSR1‐FLI1在DNA结合谱方面存在很大差异,但共享SWI/SNF相互作用。 这表明SWI/SNF结合本身可能是FET家族癌基因中重要的共同致癌机制。
图EV3。 BRG1 Co-IP评估DDIT3作为潜在的合作伙伴。
GFP标记蛋白与BRG1共免疫沉淀的免疫印迹分析(IB)。 在稳定表达FUS-DDIT3-EGFP或DDIT3-EGFP的HT1080细胞系中使用BRG1和DDIT3抗体进行检测。 为了直接量化结合蛋白和非结合蛋白的比例,将每个IP样品的相对蛋白质量加载到凝胶上,并考虑免疫沉淀过程中的稀释。 一: 输入核提取物,B:结合蛋白,NB:未结合蛋白。 显示了一个具有代表性的免疫印迹。 源数据中显示了所有复制品的免疫印迹。
显示HT1080 FUS‐DDIT3‐EGFP或DDIT3−EGFP.中DDIT3的免疫印迹结合信号强度占总(结合+非结合)信号强度的百分比的图表。 将特异性相互作用(BRG1)与非特异性相互作用(阴性对照IgG)进行比较。 平均值±SEM显示为用圆圈表示的单个重复, n个 = 3. 学生的 t吨 试验,ns=不显著。 所有量化的原始数据,包括 P(P) 值显示为源数据。
可在线获取此图的源数据。
融合癌基因FET家族的统一致病机制 FET融合癌蛋白的许多变体,除了少数例外,对每种肿瘤类型都是特定的(图 4 D) 先前的研究表明,FET癌基因对肿瘤形态和基因表达模式具有指导意义 25 , 53 从图 4 D、 很明显,转录因子伴侣及其序列特异性决定了肿瘤的类型。 Boulay报告了通过FET癌蛋白确定的基因组位点的SWI/SNF招募 等 45 我们的结果表明,所有FET‐NTD都与SWI/SNF结合,表明这种异常的SWI/SNF募集是肿瘤类型特异性背后的一个中心机制,反映了一种常见的致病机制。
恶性横纹肌样肿瘤和滑膜肉瘤分别携带BAF47/INI1/SNF5/SMARCB1和SS18 SWI/SNF核心成分的突变,从而损害SWI/SNF-功能。 这些肿瘤几乎没有其他突变,并且基因稳定 54 , 55 表明SWI/SNF突变在肿瘤发展中的中心作用。 同样,FET致癌基因引起的肿瘤基因稳定,几乎没有其他突变,重要的是,没有SWI/SNF突变的报告。 相反,我们的数据表明FET癌蛋白结合SWI/SNF并损害其功能。 FET致癌基因引起的肿瘤、恶性横纹肌样肿瘤和滑膜肉瘤的这些共同特征进一步强调了SWI/SNF和染色质重塑在癌症发展中的重要性,并指出了FET癌蛋白相关肿瘤的一种新的统一致病机制。 这种机制的靶向性可能是针对这一大类肿瘤的所有实体进行新疗法的富有成效的途径。
材料和方法 细胞培养 Raji-Burkitt淋巴瘤细胞系 56 是卡洛林斯卡研究所Georg Klein博士的一份礼物。 我们从MLS肿瘤组织中建立了黏液样脂肪肉瘤(MLS)细胞系402‐91和2645‐94 57 纤维肉瘤细胞系HT1080 58 从ATCC(CCL‐221,Manassas,VA,USA)获得,并按所述建立表达FUS‐DDIT3‐EGFP、EWSR1‐FLI1‐EGFP、DDIT3−EGFPs和EGFP的稳定克隆 59 通过检查COSMIC数据库和我们自己的蛋白质水平分析,排除了编码SWI/SNF成分的基因可能发生的突变。 除EWS细胞系外,所有细胞系均常规进行支原体感染筛查,并在RPMI1640中用谷氨酸MAX培养。 EWS细胞株TC‐71和IOR/CAR是博洛尼亚大学Katia Scotland博士赠送的礼物,并在IMDM GlutaMAX中培养。 培养基中添加5%或10%胎牛血清、100 U/ml青霉素和100μg/ml链霉素。 通过添加500μg/ml基因素维持EGFP构建物的稳定表达。 所有培养基和补充剂均来自Life Technologies(Carlsbad,CA,USA)。 细胞在含有5%CO的空气中保持在37°C 2 RT-PCR分析证实了所有使用的肉瘤细胞系的独特融合癌基因含量(图 电动汽车4 ).
图EV4。 细胞系验证。
通过对MLS 2645‐94(FUS‐DDIT3 II型)、MLS 402‐91(FUS−DDIT3I型)、EWS TC‐71(EWSR1‐FLI1 I型),EWS IOR/CAR(EWSR1-ERG)、HT1080 FUS‐的DDIT3‐EGFP(FUS‑DDIT3-II型)和HT1080 EWSR1-FLI1-EGFP的融合断点(brkp)进行RT–PCR验证细胞系:细胞系列表,融合类型, 所使用的分析(正向和反向引物)和所指示的预期产物大小。
使用引物EWSR1XhoIF:ATACTCGATGGCGCCCACGGATTACTAGCTACC和FLI1Sal1R:ATAGTCGACCGTTAGTAGTGCCTAGAGAGAGGG以与FUS‐DDIT3相同的方式构建了一个表达载体,该表达载体包含pEGFP-N1(Clontech,Mountain View,CA,USA)中EWSR1‐FLI1(type 1)的完整编码区 59 使用FuGENE瞬时转染HT1080细胞,转染pEGFP‐N1表达载体(空的或含有融合癌基因FUS‐DDIT3或EWSR1‐FLI1)或pDsRED1‐N1(克隆)表达载体(空白的或带有FUS-DDIT3,克隆方式与pEGFP-N1相同) ® 6转染试剂(美国威斯康星州麦迪逊市Promega),符合制造商的建议。 接种后第二天,细胞与转染试剂(μl)与DNA(μg)的比例为3:1,以60%的合流率进行转染。 转染dsRED/EGFP构建物24小时后制作核提取物,转染EGFP构造物24小时和48小时后制作全细胞提取物,以研究组蛋白修饰。
为了进行PLA分析,细胞生长在胶原I涂层的8孔培养玻片上(BD Biosciences,San Jose,CA,USA)。 对于tazemetostat治疗(EZH2抑制),在全细胞提取之前,将细胞接种在6孔板上,并用溶解在DMSO或DMSO对照中的5μM tazemetotstat(EPZ‐6438,Selleckchem,Munich,Germany)处理72小时。
重组蛋白表达和下拉 先前描述了编码GST融合蛋白、重组蛋白表达和纯化的载体 39 简言之,将表达载体转化为Rosetta DE3 pLysS(德国达姆施塔特诺瓦根默克公司),并用50μg/ml氨苄西林和34μg/ml氯霉素(美国密苏里州圣路易斯市西格玛奥德里奇)接种在Luria肉汤(美国加利福尼亚州圣安娜市MP Biomedicals公司)中。 细菌在37°C下隔夜培养,摇晃轨道,稀释1:20,培养至OD 600 0.6,然后通过添加最终浓度为1 mM的IPTG(默克)诱导蛋白表达。 4小时后,在6000℃离心收集细菌 克 在4°C下冷冻10分钟,在液体N中冷冻 2, 并储存在−80°C。 将颗粒解冻并重新悬浮在冰冷裂解缓冲液(50 mM NaH)中 2 人事军官 4 pH 7.5,0.5%NP‐40,300 mM NaCl),添加5 mM DTT和蛋白酶抑制剂(德国曼海姆罗氏诊断公司),然后进行超声波处理。 样品在12000℃下离心 克 在4°C下培养20分钟,上清液与预平衡的谷胱甘肽-Sepharose 4B(英国Little Chalfont的GE Healthcare)一起培养,并在4°C下旋转15分钟。 然后用洗涤缓冲液(50 mM NaH)将琼脂糖洗涤四次 2 人事军官 4 pH 7.5,1%NP‐40,500 mM NaCl,10%甘油,1 mM DTT),使用不含DTT的裂解缓冲液进行两次,并使用存储缓冲液(20 mM Tris pH 7.5,50%甘油)进行平衡。 用储存缓冲液制备25%的含有结合GST融合蛋白的琼脂糖浆液,并将其保存在−20°C。
Raji细胞在500℃离心 克 5min,用PBS清洗一次,再悬浮5–10×10 6 在冰镇蛋白提取缓冲液(20 mM Tris pH 7.5,0.5%NP–40,100 mM NaCl,2 mM MgCl)中每毫升细胞数 2 )补充1x HALT蛋白酶和磷酸酶抑制剂鸡尾酒(Pierce,Thermo Fisher Scientific,Waltham,MA,USA)。 在4°C下旋转15分钟,促进细胞溶解,然后在12000℃下离心清除细胞提取物 克 在4°C下保持20分钟。 提取对应于25–100×10 6 拉吉细胞用于每个下拉样品。 将带有结合GST融合蛋白的预平衡谷胱甘肽-Sepharose 4B(GE Healthcare)与蛋白质提取物在4°C条件下培养2-4小时。 然后用1 ml蛋白质提取缓冲液将琼脂糖洗涤四次。 固定化蛋白复合物在2x LDS样品缓冲液(Invitrogen,Thermo Fisher Scientific)中于95°C下变性10分钟洗脱。 对于具有不同NaCl浓度的实验,用含有100、150、250、500或1000mM NaCl的蛋白质提取缓冲液洗涤琼脂糖四次,然后用100mM NaCl进行一次额外的洗涤步骤,以在洗脱前平衡珠粒。 对于核酸酶的实验,在含有5 mM MgCl的改良蛋白质提取缓冲液中使用20 U/ml RNase ONE核糖核酸酶(Promega)或10 U/ml RQ1 RNase-Free DNase(Promeca) 2 ,5 mM氯化锰 2, 和10 mM CaCl 2 .样品储存在−20°C温度下。 将下拉样品置于凝胶上(见SDS–PAGE和Immunoblot),并发送合适的凝胶片进行MS分析(研究1)。
现场 近距离结扎分析和共焦成像
用PBS短暂清洗细胞,并用3.7%甲醛(Sigma‐Aldrich)在PBS中固定15分钟。 用PBS清洗样品3×10 min,并用封闭/渗透(B/P)缓冲液(PBS pH 7.3,2%BSA,0.02%Triton X‐100;均来自Sigma‐Aldrich)培养30 min。 然后将样品与一级抗体组合培养90分钟:1μg/ml ARID1A(HPA005456;Sigma‐Aldrich)、4μg/ml BRG1(H88)(sc‐10768;Santa Cruz Biotechnology,Santa Cru z,CA,USA)、3μg/ml DDIT3(9C8)(ab11419;Abcam,Cambridge,United Kingdom)、稀释在B/P缓冲液中的2μg/ml FUS(4H11)(sc‐47711;Santa Cruz)。 通过省略两对一级抗体中的一个,进行了对照实验。 在PBS中用0.1%吐温-20温和搅拌3×5 min冲洗载玻片。 现场 用Duolink进行邻近连接分析(PLA) 现场 荧光产品(OLink,乌普萨拉,瑞典)符合制造商的说明。 简单地说,样品在37°C下与PLA探针抗兔PLUS和抗鼠MINUS的混合物孵育1小时,每个探针在B/P缓冲液中稀释1:5。 将载玻片在1x洗涤缓冲液A(OLink)中洗涤2×5 min,然后用Duolink进行结扎、洗涤步骤和扩增 现场 检测试剂红(OLink)是根据制造商的说明制备的。 使用Duolink用盖玻片安装幻灯片 现场 使用DAPI(OLink)装载介质。 采用带有LSM-5软件的蔡司LSM510 META共焦显微镜系统(德国奥伯科钦蔡司)进行共焦成像。 使用63x/1.4油物镜和顺序扫描,具有适合每个荧光团的激发和META检测器过滤器设置(激发561 nm和BP600‐710用于PLA信号,激发405 nm和BP420‐475用于DAPI)。
核蛋白提取和免疫共沉淀 从两个融合的T75或15厘米培养皿中收集细胞,在PBS(生命技术)中刮取,然后在450℃离心 克 在4°C下保持10分钟。 将细胞颗粒(约100–200μl填充细胞体积)重新悬浮在5个填充细胞体积的低渗裂解缓冲液(10 mM KCl,10 mM Tris pH 7.5,1.5 mM MgCl)中 2 ; 所有Life Technologies)补充1 mM DTT(Sigma‐Aldrich)和1x Halt蛋白酶抑制剂鸡尾酒(Thermo Scientific、Thermo Fisher Scientistic),并在冰上膨胀15分钟。 在400℃离心后,将上清液丢弃 克 在4°C下保持5分钟。 将填充好的细胞重新悬浮在2个填充细胞体积的低渗裂解缓冲液中,并用带有27号针头的注射器进行2至5次冲程。 在10000离心后去除细胞质部分 克 在4°C下保持20分钟。 然后将球粒细胞核重新悬浮在(2/3)填充细胞体积的高盐提取缓冲液中[0.42 M KCl,10 mM Tris pH 7.5,0.1 mM EDTA(所有生命科技公司),10%甘油(默克化学公司,默克公司)]补充1x Halt蛋白酶抑制剂鸡尾酒,并在冰箱中轻轻搅拌30分钟。 在20000离心后收集核部分 克 在4°C下保持5分钟,并稀释至150 mM盐浓度。 在Brg1 IP定量实验、DDIT3 IP和FLI1 IP的细胞破碎过程中,加入苯甲酸酶(#71205,Merck Millipore,Merck),在4°C下用5 U/ml处理15分钟。
使用适用于下游质谱分析的方案,用Dynabeads Myone Streptavidin T1(Thermo Fisher Scientific)免疫沉淀核提取物。 用补充有1x Halt蛋白酶抑制剂Cocktail的IP洗涤缓冲液(150 mM KCL,10 mM Tris pH 7.5,0.1 mM EDTA,10%甘油)将核提取物(60μl,100–200μg)稀释至500μl,并与10μg抗体,Brg1‐生物素(ab200911,Abcam)或正常小鼠IgG生物素(sc‐2762,Santa Cruz Biotechnology)混合。 核提取物/抗体混合物在4°C温和旋转培养过夜。 第二天,将75μl珠子(每个反应)在Rotiblock(Carl Roth,Karlsruhe,Germany)中封闭约30分钟,然后用IP洗涤缓冲液进行三次洗涤。 然后将核提取物/抗体混合物添加到珠子中,并在4°C下轻轻旋转培养2小时。 将珠粒用10mM TEAB缓冲液(Thermo Fisher Scientific)轻轻旋转洗涤3×5分钟。 用20μl 1%甲酸(Sigma‐Aldrich)在50°C,500 rpm的条件下两次洗脱捕获的蛋白质复合物5分钟,然后用20μl 2x LDS样品缓冲液和10%样品还原剂(Life Technologies)在90°C,500rpm的条件下第三次洗脱10分钟,以确保释放所有结合蛋白。 凝胶分析后,将甲酸洗脱液送往MS分析(研究2)。 对于Brg1 IP定量实验,使用100μg核提取物,洗脱前的最后三次洗脱在IP洗脱缓冲液中进行,样品在含有10%样品还原剂的2×50μl 2x LDS样品缓冲液中洗脱。 BRG1 IP复制被缩小了两倍。 对于DDIT3 IP(DDIT3生物素抗体,NB600‐1335B,Novus Biologicals,Littleton,CO,USA)和FLI1 IP(FLI1生物素抗体、US生物素246159生物素,Salem,MA,USA,),该方案扩大了三倍,样品在2×75μl或2×150μl 2x LDS样品缓冲液中用10%样品还原剂洗脱。 凝胶分析后,将DDIT3 IP(研究3)和FLI1 IP(研究4)的LDS洗脱液送往MS分析。 对于定量实验,将每个IP样品的相对数量的蛋白质加载到凝胶上,并考虑免疫沉淀过程中的稀释,以便直接定量结合蛋白和非结合蛋白的分数。
质谱法 蛋白质组学分析在哥德堡大学Sahlgrenska学院的蛋白质组学核心设施进行。凝胶片(研究1)用25 mM碳酸氢铵在50%乙腈(ACN)中脱色,通过在50 mM碳铵中添加10 ng/μl胰蛋白酶(Pierce MS级,Thermo Fisher Scientific)进行凝胶内消化, 并在37°C下孵育过夜。 用50%ACN和1%乙酸从凝胶中提取肽并干燥。 使用过滤辅助样品制备(FASP)方法用胰蛋白酶消化免疫沉淀(IP)中的甲酸(研究2)和LDS(研究3和4)洗脱液 60 简单地说,用100 mM二硫苏糖醇在60°C下还原样品30分钟,转移到30 kDa MWCO Pall Nanosep离心过滤器(Sigma‐Aldrich),用8 M尿素反复洗涤,并用10 mM甲基甲烷硫磺酸盐烷基化。 通过添加0.30μg Pierce MS级胰蛋白酶,在37°C的50 mM TEAB、1%脱氧胆酸钠(SDC)缓冲液中进行消化,并培养过夜。 添加额外部分胰蛋白酶并再培养2小时。 通过离心收集肽,用10%三氟乙酸酸化去除SDC。 根据制造商的指南,使用PepClean C18旋转柱(赛默飞世尔科技公司)对样品进行脱盐,并进行干燥。 研究4中的样品在C18脱盐之前通过HiPPR洗涤剂去除旋转柱(赛默飞世尔科技公司)进行处理。 样品在3%ACN和0.2%甲酸(FA)中重新配制。
肽样品在配有7T ICR磁铁(LTQ‐FT,研究1)、Orbitrap Fusion Tribrid质谱仪(研究2和3)或Q Exactive HF(研究4)质谱仪(均为Thermo Fisher Scientific)的混合线性离子阱-FTICR质谱仪上进行分析,该质谱仪与Easy nLC 1000液相色谱系统接口。 使用C18预柱(45×0.075 mm I.D.)和分析柱(300×0.075 mmI.D.)捕获并分离肽,分析柱填充3μm Reprosil‐Pur C18‐AQ颗粒,梯度为5至80%ACN,置于0.2%FA中40分钟。 MS光谱和MS/MS光谱分别在FTICR和LTQ阱中获得。 在FTICR的每次扫描中,三个最强烈的双电荷或三电荷离子通过碰撞诱导解离(CID)在线性陷阱中顺序破碎。 在研究2和3中,以120000分辨率获取前体离子质谱,并以数据相关模式进行MS/MS分析,其中最强烈的前体离子的CID光谱以30000分辨率记录在离子阱中,碰撞能量设置为30持续3秒(“最高速度”设置)。 选择电荷状态2–7进行碎片化,动态排除设置为45秒。 在研究4中,以60000分辨率获取前体离子质谱,并以数据相关模式进行MS/MS分析,其中前10个最强前体离子的HCD光谱以30000分辨率记录。 选择电荷态2–4进行碎片化,碰撞能量设置为28,动态排除设置为20秒。
利用Proteome Discoverer 1.4版(Thermo Fisher Scientific)对2009年55.3版的瑞士人数据库(研究1,与2017年11月的版本进行验证以进行数据上传)、2016年5月(研究2)、9月(研究3)和2017年3月(研究4)进行数据分析。 吉祥物(2.3或2.5.1,Matrix Science)用作搜索引擎,前体质量容差为5 ppm,碎片质量容差500 mmu。 色氨酸肽通过一次缺失裂解、可变蛋氨酸氧化和静态半胱氨酸丙酰胺修饰(研究1)或零到一次缺失的裂解和可变蛋氨酸氧化、静态半胱胱氨酸甲硫代修饰(研究2、3和4)被接受。 通过搜索反向数据库,将软件中检测到的肽阈值设置为马斯科95%(研究1)或马斯科99%(研究2、3和4)的显著性水平,并通过共享相同序列对已识别的蛋白质进行分组,以最小化冗余。
组蛋白修饰分析的全细胞提取 在冰上制备全细胞提取物,用70%至95%的10厘米或15厘米培养皿(稳定克隆)或6孔培养板(瞬时转染后)融合细胞,在PBS中刮擦,然后在450℃离心 克 在4°C下保持10分钟。 将细胞沉淀在含有5mM EDTA和1x Halt蛋白酶抑制剂鸡尾酒的RIPA缓冲液(25mM Tris•HCl pH 7.6,150mM NaCl,1%NP‐40,1%脱氧胆酸钠,0.1%十二烷基硫酸钠,Pierce,Thermo Scientific)中裂解,并在冰上孵育10分钟,每5分钟轻轻混合一次。 然后对裂解液进行超声波处理,以破坏粘性DNA。 为了不丢失不溶性部分中残留的组蛋白,此时未进行离心。 然后将样品与NuPAGE 4x LDS样品缓冲液混合至最终浓度106 mM Tris–HCl、141 mM Tris-Base、2%LDS、10%甘油、0.51 mM EDTA、0.22 mM SERVA Blue G250和0.175 mM酚红,pH 8.5,并在95°C下加热10分钟,然后装入凝胶。 用免疫印迹分析等量的蛋白质,以评估组蛋白3赖氨酸4三甲基化(H3K4me3)、组蛋白3赖氨酸27三甲基化(H3K27me3)和组蛋白3赖氨酸27乙酰化(H3K27Ac)的量,使用组蛋白H4作为负载对照,见免疫印迹。
SDS-PAGE和免疫印迹 根据制造商的说明,使用Novex NuPAGE系统(生命科技公司)用SDS–PAGE对蛋白质样品进行粒径分离。 简而言之,蛋白质提取物与1x NuPAGE LDS样品缓冲液和10%NuPAGE样品还原剂混合,在70°C下变性10分钟,并在NuPAGE 4–12%Bis‐Tris或3–8%Tris‐醋酸盐凝胶上分离。 分离的蛋白质用SimpleBlue SafeStain(Life Technologies)染色或通过湿印迹转移到聚偏二氟乙烯膜(0.45μm,Life Technols)上。 在TBS‐T缓冲液(50 mM Tris‐HCl pH 6.8,50 mM NaCl,0.1%吐温‐20;均来自Sigma‐Aldrich)中用5%脱脂牛奶(默克化学)或5%BSA(西格玛‐奥尔德里奇)封闭膜。 将膜在4°C下与0.5μg/ml ARID1A(PSG3)(sc‐32761;Santa Cruz)、0.5μg/ml ARID1A, 0.5μg/ml BRG1(H‐88)(sc‐10768;圣克鲁斯)、0.2μg/ml BREG1(g‐7)(sc-17796;圣克鲁兹)、0.7μg/ml DDIT3), 1μg/ml GAPDH(ab9484;Abcam)、0.5μg/ml GFP(JL‐8)(632381;Clontech)、1μg/ml H3K4me3(#05‐745R;Merck Millipore)、0.1μg/ml H3K27ac(ab177178;Abcam)、0.5微克H3K17me3(#07‐449;Merck Millipore; Santa Cruz),然后在室温下与抗小鼠或抗兔HRP偶联的第二抗体(32430和32460;Thermo Scientific)孵育1个小时。 在与SuperSignal West Dura Extended Dura基板或SuperSignal West Femto Max Sensitivity基板(Thermo Scientific)孵育后,使用ImageQuant LAS 4000 mini(GE Healthcare Life Sciences)通过发光信号捕获蛋白质检测。 在室温下培养15分钟,用ReBlot Plus(2504,Merck Millipore)剥离一些膜,然后用另一种一级抗体重新标记膜。 使用MultiGauge V3.2(日本东京富士胶片公司)对色带进行量化。
统计分析 3–4个重复的免疫印迹定量值表示为平均值±SEM,每个单独的实验都表明。 统计分析使用GraphPad Prism软件(7.00版,美国加利福尼亚州圣地亚哥GraphPat)。 学生的 t吨 测试(未配对,双面 t吨 ‐test),α<0.05显著。 所有量化的原始数据都显示在源数据中。
RNA‐测序 Smart‐seq2协议 61 用于从HT1080 wt生成测序库( n个 =4),HT1080 EGFP( n个 =4),HT1080保险丝‐DDIT3‐EGFP( n个 =3)和HT1080 EWSR1‐FLI1‐EGFP( n个 = 4). 用DPBS清洗粘附细胞,并直接在添加了β‐巯基乙醇(MP生物医学)的RLT裂解缓冲液(德国希尔登市齐根)中刮取。 根据制造商的建议,使用RNeasy Micro Kit和DNase处理(Qiagen)提取总RNA,并将其储存在−80°C下。 在2100生物分析仪上使用安捷伦RNA 6000纳米试剂盒(安捷伦科技,加利福尼亚州圣克拉拉,美国)确认RNA质量。
对10 ng总RNA进行反转录。通过向样品中添加1 mM dNTP和1μM生物素化适配器序列(含寡聚-dT30VN(5′-生物素-AAGCAGTATCAACGCAGTACT30VN‐3′)来执行初始杂交步骤(Sigma‐Aldrich浓度均指最终的反转录反应) 并在72°C下孵育3分钟。 随后,1x第一链缓冲液(50 mM Tris–HCl pH 8.3、75 mM KCl和3 mM MgCl 2 ),5 mM二硫苏糖醇(均为Invitrogen,Thermo Fisher Scientific),10 mM MgCl 2 (Ambion,Thermo Fisher Scientific),1 M甜菜碱(Sigma‐Aldrich),0.6μM生物素化适配器序列包含模板切换寡核苷酸(5′-生物素‐AAGCAGTGATCAACGCAGTACATrGrG+G‐3′,rG=核糖鸟嘌呤和+G=锁定核酸修饰鸟嘌呤s,Eurogentec,Liège,Belgien),15 U RNaseOUT, 和150U SuperScript II(均为Invitrogen,Thermo Fisher Scientific),得到15μl的反应体积。 在T100仪器中,在42°C下逆转录90分钟,在70°C下反转录15分钟(美国加利福尼亚州赫拉克勒斯市Bio‐Rad)。 cDNA储存于−20°C。
将7.5μl cDNA样品与1x KAPA Hifi HotStart Ready Mix(KAPA Biosystems,Wilmington,MA,USA)、0.1μM引物(5′-AAGCAGTATCAACGCAGT‐3′;Sigma‐Aldrich)混合,反应体积为50μl,进行预扩增。 预扩增在98°C下进行3分钟,然后在98°C下进行24个扩增循环20秒,67°C下进行15秒,72°C下进行6分钟,最后在T100仪器中在72°C下再孵育5分钟。 样品从72°C直接转移到干冰中,并储存在−20°C下。 使用Agencourt AMPure XP珠(BD Biosciences)纯化样品。
将50μl样品与40μl珠子(珠子与样品的比例为0.8)混合,然后在室温下在工作台上培养5分钟,在磁铁上培养5 min(DynaMag,Thermo Fisher Scientific)。 丢弃上清液,用200μl 80%乙醇清洗珠子两次,然后晾干。 使用17.5μl不含RNase/DNase的水(Invitrogen,Thermo Fisher Scientific)在室温下在工作台上培养2 min,在磁铁上培养2 min.对样品进行洗脱。 使用安捷伦高灵敏度DNA试剂盒在2100生物分析仪(安捷伦科技)上进行质量和浓度测量,并使用100 pg的cDNA进行标记和索引。
使用Nextera XT DNA文库准备试剂盒和Nextera-XT索引试剂盒v2(Illumina,San Diego,CA,USA)进行标记和索引。 首先,向5μl样品中添加10μl Tagment DNA缓冲液和5μl Amplicon Tagment Mix,并在T100仪器中在55°C下进行5分钟的标记。 接下来,加入5μl中和标记缓冲液,然后在1100 rpm下离心1分钟(LMC‐3000,转子R‐2,Biosan,里加,拉脱维亚),并在室温下孵育5分钟。 对于索引和文库扩增,添加15μl Nextera PCR Master Mix和5μl每个索引1(i7)和索引2(i5)适配器,并在72°C下扩增3分钟,95°C扩增30秒,然后在95°C下16次扩增10秒,55°C扩增30s,72°C扩增30ms, 并在T100仪器中在72°C下进行最后一次额外培养5分钟。 与之前一样,使用Agencourt AMPure XP珠进行样品纯化,但使用0.6的珠样比,将所有样品体积添加到30μl珠中。
使用Qubit dsDNA高灵敏度分析试剂盒(Invitrogen,Thermo Fisher Scientific)分析每个样品的浓度。 使用DNF‐474高灵敏度NGS试剂盒(两种安捷伦技术)在片段分析仪上通过毛细管凝胶电泳确保文库质量控制和大小。 根据片段分析仪数据对文库进行等摩尔混合,并使用NEBNext Library Quantification kit(美国马萨诸塞州伊普斯威奇新英格兰生物实验室)通过qPCR对最终文库进行量化。 在MiniSeq仪器(均为Illumina)上,利用读取长度为2×75 bp的配对末端测序,以1.8 pM的浓度对文库进行聚类,并辅以1%PhiX对照。
使用STAR RNA‐seq校准器v2.6校准Illumina读数 62 使用ENSEMBL GRCh38组合作为参考基因组。 读取计数矩阵是使用HTSeq python框架v0.9.1生成的。 63 总计数<10的基因被排除在下游分析之外。 使用R包DESeq2分析差异表达,基于负二项分布模型对弥散度和折叠变化的收缩估计 64 .已调整 P(P) 使用Benjamini–Hochberg方法计算值。 基因至少双重调节(调整 P(P) ‐值≤0.05),在下游分析中使用分子特征数据库(MSigDB)v6.2进行分析 65 , 66 将基因列表与基因集集合“化学和遗传扰动”(GSEA: http://software.broadinstitute.org/gsea/msigdb/index.jsp )和前20个基因集(FDR q个 ‐值<0.05)。
作者贡献 ML和CT参与了主要的实验室工作,计划和设计了实验,准备了数字和其他结果演示,并主要参与了手稿的撰写。 PG进行了组蛋白分析和相关的细胞培养/克隆工作,并编写了相应的手稿部分。 EJ帮助进行细胞培养工作、数据处理和格式化,并帮助撰写手稿。 DA和SD参与了手稿撰写和图形制作; RR做了重要的实验室工作,并帮助撰写了手稿。 简历中添加了EZH2分析,并帮助撰写了手稿。 MLS帮助对RNA‐seq数据进行生物信息学分析。 HF和AS对关键数据的解释和手稿的撰写做出了贡献。 PÅ参与了项目规划、实验设计、数据处理和解释以及手稿的撰写。
致谢 本研究得到了瑞典癌症协会(CAN2015/7130,2016/438)、瑞典政府与县议会协议下的瑞典州、ALF协议(ALFGBG‐722211和716321)、克努特和爱丽丝·沃伦伯格基金会、哥德堡大学沃伦伯格分子和转化医学中心、, 瑞典研究理事会(2017‐01392)、VINNOVA、瑞典医学研究学会、阿萨·加布里尔森基金会、BioCARE国家战略研究、瑞典儿童癌症基金会(PR2017‐0043)、约翰·詹森癌症研究基金会、瑞典医学会, 以及威廉和玛蒂娜·伦德格伦科学研究基金会。 我们感谢哥德堡大学Sahlgrenska学院的细胞成像中心在成像方面提供的帮助,感谢#explanartist的Jacqueline Forzelius提供的图形演示。 哥德堡大学Sahlgrenska学院的蛋白质组学核心设施进行了蛋白质鉴定的质谱分析。 我们感谢Inga‐Britt和Arne Lundbergs Forskningsstiftelse捐赠Orbitrap Fusion Tribrid MS仪器。
参与者信息 Anders Stáhlberg,电子邮件: anders.stahlberg@gu.se。
皮埃尔·奥曼,电子邮件: pierre.aman@gu.se。
参考文献
1 Hoell JI、Larsson E、Runge S、Nusbaum JD、Duggimpudi S、Farazi TA、Hafner M、Borkhardt A、Sander C、Tuschl T(2011)野生型和突变FET家族蛋白质的RNA靶点。 自然结构分子生物学 18: 1428–1431 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
2 Ishigaki S、Masuda A、Fujioka Y、Iguchi Y、Katsuno M、Shibata A、Urano F、Sobue G、Ohno K(2012)位置依赖型FUS‐RNA相互作用调节选择性剪接事件和转录。 科学代表 2: 529 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
三。 Meissner M、Lopato S、Gotzmann J、Sauermann G、Barta A(2003)原癌蛋白TLS/FUS与核基质相关,并与剪接因子PTB、SRm160和SR蛋白复合。 Exp单元Res 283: 184–195 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
4 Yang L,Embree LJ,Tsai S,Hickstein DD(1998)癌蛋白TLS与参与RNA剪接的丝氨酸精氨酸蛋白相互作用。 生物化学杂志 273: 27761–27764 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
5 Zinszner H,Sok J,Immanuel D,Yin Y,Ron D(1997)TLS(FUS)结合RNA 体内 并参与核质穿梭。 细胞科学杂志 110(第15部分):1741-1750 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
6 Fujii R,Takumi T(2005)TLS促进编码肌动蛋白稳定蛋白的mRNA向树突棘的转运。 细胞科学杂志 118: 5755–5765 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
7 Gregory RI、Yan KP、Amuthan G、Chendrimada T、Doratotaj B、Cooch N、Shiekhattar R(2004)微处理器复合体介导微RNA的生成。 自然 432: 235–240 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
8 奥曼·P(1999)《实体肿瘤中的融合基因》。 赛明癌症生物学 9: 303–318 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
9 Riggi N、Cironi L、Suva ML、Stamenkovic I(2007)《肉瘤:遗传学、信号传递和细胞起源》。 第一部分:TET联谊会。 病理学杂志 213: 4–20 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
10 Perez‐Mancera PA,Sanchez‐Garcia I(2005)《了解间叶癌:脂肪肉瘤相关FUS‐DDIT3融合基因作为模型》。 赛明癌症生物学 15: 206–214 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
11 Riggi N,Stamenkovic I(2007)《尤文肉瘤生物学》。 癌症快报 254: 1–10 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
12 Riggi N、Cironi L、Provero P、Suva ML、Kaloulis K、Garcia‐Echeverria C、Hoffmann F、Trumpp A、Stamenkovic I(2005)《从原始骨髓源性间充质祖细胞发展尤因肉瘤》。 Can Res公司 65: 11459–11468 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
13 Stáhlberg A,Kábjörn Gustafsson C,Engström K,Thomsen C,Dolatabadi S,Jonasson E,Li CY,Ruff D,Chen SM,O-man P(2014)遗传稳定黏液/圆细胞脂肪肉瘤中的正常和功能TP53。 公共科学图书馆 9:e113110 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
14 Crompton BD、Stewart C、Taylor‐Weiner A、Alexe G、Kurek KC、Calicchio ML、Kiezun A、Carter SL、Shukla SA、Mehta SS 等 (2014)儿童尤因肉瘤的基因组景观。 癌症发现 4: 1326–1341 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
15 Tirode F、Surdez D、Ma X、Parker M、Le Deley MC、Bahrami A、Zhang Z、Lapouble E、Grossetete‐Lalami S、Rusch M 等 (2014)尤因肉瘤的基因组景观定义了一种侵袭性亚型,与STAG2和TP53突变相关。 癌症发现 4: 1342–1353 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
16 Ng KP、Potikyan G、Savene RO、Denny CT、Uversky VN、Lee KA(2007),无序结构中的多个芳香侧链对EWS家族癌蛋白的转录和转化活性至关重要。 《美国科学院院刊》 104: 479–484 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
17 奥曼P(2005)肿瘤发展中的融合癌基因。 赛明癌症生物学 15:236–243 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
18 Bailly RA、Bosselut R、Zucman J、Cormier F、Delatter O、Roussel M、Thomas G、Ghysdael J(1994),尤因肉瘤t(11;22)易位导致的EWS‐FLI‐1融合蛋白的DNA结合和转录激活特性。 分子细胞生物学 14: 3230–3241 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
19 Zinszner H,Albalat R,Ron D(1994)CHOP的致癌转化需要来自RNA结合蛋白TLS或EWS的新效应结构域。 基因开发 8: 2513–2526 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
20 Bertolotti A,Bell B,Tora L(1999)人类TAFII68的N末端结构域显示了转录激活和致癌特性。 癌基因 18: 8000–8010 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
21 Petermann R、Mossier BM、Aryee DN、Khazak V、Golemis EA、Kovar H(1998)致癌EWS‐Fli1与人类RNA聚合酶II的亚单位hsRPB7相互作用。 癌基因 17: 603–610 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
22 Powers CA、Mathur M、Raaka BM、Ron D、Samuels HH(1998)TLS(脂肪肉瘤易位)是类固醇、甲状腺激素和维甲酸受体的高亲和力相互作用物。 分子内分泌学 12: 4–18 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
23 Ohno T,Rao VN,Reddy ES(1993)EWS/Fli‐1嵌合蛋白是一种转录激活物。 Can Res公司 53: 5859–5863 [ 公共医学 ] [ 谷歌学者 ]
24 Tomazou EM、Sheffield NC、Schmidl C、Schuster M、Schonegger A、Datlinger P、Kubicek S、Bock C、Kovar H(2015)表观基因组定位揭示了致癌融合蛋白EWS‐FLI1的不同基因调控模式和广泛增强子重编程。 单元格代表 10: 1082–1095 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
25 Engström K,Willen H,Kábjörn‐Gustafsson C,Andersson C,Olsson m,Göransson m,Järnum S,Olofson A,Warnhammar E,Oman P(2006)黏液/圆细胞脂肪肉瘤融合癌基因FUS‐DDIT3和正常DDIT3在转染的人纤维肉瘤细胞中诱导脂肪肉癌表型。 美国病理学杂志 168: 1642–1653 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
26 Sheffield NC、Pierron G、Klughammer J、Datlinger P、Schonegger A、Schuster M、Hadler J、Surdez D、Guillemot D、Lapouble E 等 (2017)DNA甲基化异质性定义了尤因肉瘤的疾病谱。 自然·医学 23: 386–395 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
27 Svoboda LK、Harris A、Bailey NJ、Schwentner R、Tomazou E、von Levetzow C、Magnuson B、Ljungman M、Kovar H、Lawlor ER(2014)HOX基因的过度表达在尤文肉瘤中普遍存在,并与发育转录程序表观遗传调控的改变有关。 表观遗传学 9: 1613–1625 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
28 奥曼·P、帕纳戈普洛斯一世、拉森·C、菲奥雷托斯·T、门辛格·M、托雷斯·H、霍格伦德·M、福斯特·A、拉比特斯·T、罗恩·D 等 (1996)人类肉瘤相关基因FUS和EWS的表达模式以及FUS的基因组结构。 基因组学 37:1–8 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
29 Morohoshi F、Arai K、Takahashi EI、Tanigami A、Ohki M(1996)克隆和定位人类RBP56基因,该基因编码与FUS/TLS和EWS蛋白类似的假定RNA结合蛋白。 基因组学 38: 51–57 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
30 Tan AY,Manley JL(2009)蛋白质的TET家族:在疾病中的功能和作用。 分子细胞生物学杂志 1:82–92 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
31 Thomsen C、Grundevik P、Elias P、Stáhlberg A、Auman P(2013)FUS、EWSR1、TAF15及其致癌融合蛋白之间的复合物形成需要保守的N末端基序。 美国财务会计准则委员会J 27: 4965–4974 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
32 Panagopoulos I,Hoglund M,Mertens F,Mandahl N,Mitelman F,Aman P(1996)黏液样脂肪肉瘤中EWS和CHOP基因的融合。 癌基因 12: 489–494 [ 公共医学 ] [ 谷歌学者 ]
33 Kadoch C、Williams RT、Calarco JP、Miller EL、Weber CM、Braun SM、Pulice JL、Chory EJ、Crabtree GR(2017)正常和致癌状态下异染色质上BAF‐多梳复合物对抗的动力学。 自然基因 49: 213–222 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
34 Stanton BZ、Hodges C、Calarco JP、Braun SM、Ku WL、Kadoch C、Zhao K、Crabtree GR(2017)Smarca4 ATP酶突变破坏PRC1从染色质中的直接驱逐。 自然基因 49: 282–288 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
35 Kadoch C、Hargreaves DC、Hodges C、Elias L、Ho L、Ranish J、Crabtree GR(2013)哺乳动物SWI/SNF复合物的蛋白质组学和生物信息学分析确定了其在人类恶性肿瘤中的广泛作用。 自然基因 45: 592–601 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
36 Andersson MK、Stáhlberg A、Arvidsson Y、Olofsson A、Semb H、Stenman G、Nilsson O、Auman P(2008)多功能FUS、EWS和TAF15原癌蛋白显示细胞类型特异性表达模式,并参与细胞扩散和应激反应。 BMC细胞生物学 9: 37 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
37 Hicks GG、Singh N、Nashabi A、Mai S、Bozek G、Klewes L、Arapovic D、White EK、Koury MJ、Oltz EM 等 (2000)小鼠Fus缺乏会导致B淋巴细胞发育和活化缺陷、染色体高度不稳定和围产期死亡。 自然基因 24: 175–179 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
38 Morohoshi F、Ootsuka Y、Arai K、Ichikawa H、Mitani S、Munakata N、Ohki M(1998)人类RBP56/hTAFII68和FUS/TLS基因的基因组结构。 基因 221: 191–198 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
39 Thomsen C、Udhane S、Runnberg R、Wiche G、Stáhlberg A、Au man P(2012)《肉瘤融合(FUS)与细胞连接蛋白plectin的相互作用:FUS亚细胞定位和功能的意义》。 Exp单元Res 318: 653–661 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
40 奥曼·P、杜拉塔巴迪·S、Svec D、Jonasson E、Safavi S、Andersson D、Grundevik P、Thomsen C、Stóhlberg A(2016)脂肪肉瘤中FUS‐DDIT3融合癌基因的调控机制、表达水平和增殖效应。 病理学杂志 238: 689–699 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
41 Eukilchen GM、Auerbach RK、Davidov E、Gianoulis TA、Zhong G、Rozowsky J、Bhardwaj N、Gerstein MB、Snyder M(2011)使用全球方法揭示SWI/SNF染色质重塑复合体的不同作用和相互作用。 公共科学图书馆-基因 7:e1002008 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
42 Kadoch C,Crabtree GR(2013)滑膜肉瘤中SS18-SSX致癌融合对mSWI/SNF(BAF)复合物的可逆破坏。 单元格 153: 71–85 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
43 Thaete C、Brett D、Monaghan P、Whitehouse S、Rennie G、Rayner E、Cooper CS、Goodwin G(1999)SYT和SYT‐SSX滑膜肉瘤易位蛋白的功能域以及与核内SNF蛋白BRM的共定位。 人类分子遗传学 8:585–591 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
44 Wilson BG、Wang X、Shen X、McKenna ES、Lemieux ME、Cho YJ、Koellhoffer EC、Pomeroy SL、Orkin SH、Roberts CW(2010)致癌转化过程中多梳和SWI/SNF复合物之间的表观遗传拮抗。 癌细胞 18: 316–328 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
45 Boulay G、Sandoval GJ、Riggi N、Iyer S、Buisson R、Naigles B、Awad ME、Rengarajan S、Volorio A、McBride MJ 等 (2017)通过朊蛋白样结构域对BAF复合物进行癌症特异性重定位。 单元格 171:163–178 e119 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
46 May WA、Gishizky ML、Lessnick SL、Lunsford LB、Lewis BC、Delatter O、Zucman J、Thomas G、Denny CT(1993)《尤因肉瘤11》; 22易位产生嵌合转录因子,需要FLI1编码的DNA结合域进行转化。 《美国科学院院刊》 90:5752–5756 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
47 Ferreira M、Verbinnen I、Fardilha M、Van Eynde A、Bollen M(2018)睾丸中蛋白磷酸酶1调节物NIPP1的缺失导致组蛋白甲基转移酶EZH2的过度磷酸化和降解。 生物化学杂志 293: 18031–18039 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
48 Ron D,Habener JF(1992)CHOP,一种新的发育调节核蛋白,与转录因子C/EBP和LAP二聚体,作为基因转录的显性负抑制剂发挥作用。 基因开发 6: 439–453 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
49 Jauhiainen A、Thomsen C、Strömbom L、Grundevik P、Andersson C、Danielsson A、Anderson MK、Nerman O、Rörkvist L、Stöhlberg A 等 (2012)应激诱导蛋白DDIT3/CHOP/GADD153的不同细胞质和核功能。 公共科学图书馆 7:e33208 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
50 韩J、巴克·SH、胡J、林YH、吉尔德斯维尔R、单J、袁CL、克罗科斯基D、王S、哈佐格鲁M 等 (2013)内质网应激诱导的转录调控增加蛋白质合成,导致细胞死亡。 Nat细胞生物学 15: 481–490 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
51 Bilke S、Schwentner R、Yang F、Kauer M、Jug G、Walker RL、Davis S、Zhu YJ、Pineda M、Meltzer PS 等 (2013)癌基因ETS融合解除了尤文肉瘤和前列腺癌中E2F3靶基因的调控。 基因组研究 23: 1797–1809 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
52 Guillon N、Tirode F、Boeva V、Zynovyev A、Barillot E、Delatter O(2009)致癌EWS‐FLI1蛋白结合 体内 具有潜在转录激活功能的GGAA微卫星序列。 公共科学图书馆 4:e4932 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
53 Hu‐Lieskovan S,Zhang J,Wu L,Shimada H,Schofield DE,Triche TJ(2005)EWS‐FLI1融合蛋白上调神经嵴发育中的关键基因,并负责观察到的尤因家族肿瘤的表型。 Can Res公司 65: 4633–4644 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
54 Limon J、Mrozek K、Mandahl N、Nedoszytko B、Verest A、Rys J、Niezabitowski A、Babinska M、Nosek H、Ochalek T 等 (1991)滑膜肉瘤的细胞遗传学:10例新病例的介绍和文献综述。 基因染色体癌 3: 338–345 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
55 Hasselblatt M、Isken S、Linge A、Eikmeier K、Jeibmann A、Oyen F、Nagel I、Richter J、Bartelheim K、Kordes U 等 (2013)高分辨率基因组分析表明,在非典型畸胎瘤/横纹肌瘤中,除SMARCB1异常外,没有复发性基因组改变。 基因染色体癌 52: 185–190 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
56 Pulvertaft JV(1965)《尼日利亚恶性肿瘤短期组织培养研究》。 临床病理学杂志 18: 261–273 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
57 奥曼·P、罗恩·D、曼达尔·N、菲奥雷托斯·T、海姆·S、阿赫登·K、威伦·H、瑞德霍尔姆·A、米特尔曼·F(1992)黏液样脂肪肉瘤中转录因子基因CHOP与T(12;16)(q13;p11)的重排。 基因染色体癌 5:278–285 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
58 Rasheed S、Nelson‐Rees WA、Toth EM、Arnstein P、Gardner MB(1974)新衍生人类肉瘤细胞系的特征(HT‐1080)。 癌症 33: 1027–1033 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
59 Thelin‐Járnum S,Göransson M,Burguete AS,Olofsson A,Au man P(2002)黏液性脂肪肉瘤特异性TLS‐CHOP融合蛋白定位于不同于PML核小体的核结构。 国际癌症杂志 97: 446–450 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
60 Wisniewski JR,Zougman A,Nagaraj N,Mann M(2009)蛋白质组分析通用样品制备方法。 Nat方法 6: 359–362 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
61 Picelli S、Faridani OR、Bjorklund AK、Winberg G、Sagasser S、Sandberg R(2014)使用Smart‐seq2从单个细胞中提取全长RNA‐seq。 Nat协议 9: 171–181 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
62 Dobin A、Davis CA、Schlesinger F、Drenkow J、Zaleski C、Jha S、Batut P、Chaisson M、Gingeras TR(2012)STAR:超快通用RNA‐seq比对仪。 生物信息学 29: 15–21 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
63 Anders S、Pyl PT、Huber W(2015)HTSeq–一个用于处理高通量测序数据的Python框架。 生物信息学 31: 166–169 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
64 Love MI、Huber W、Anders S(2014),利用DESeq2对RNA‐seq数据的折叠变化和离散度进行适度估计。 基因组生物学 15: 550 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
65 Subramanian A、Tamayo P、Mootha VK、Mukherjee S、Ebert BL、Gillette MA、Paulovich A、Pomeroy SL、Golub TR、Lander ES 等 (2005)基因集富集分析:解释全基因组表达谱的基于知识的方法。 《美国科学院院刊》 102: 15545–15550 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
66 Liberzon A、Subramanian A、Pinchback R、Thorvaldsdóttir H、Tamayo P、Mesirov JP(2011)分子签名数据库(MSigDB)3.0。 生物信息学 27: 1739–1740 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
67 Perez‐Riverol Y、Csordas A、Bai J、Bernal‐Llinares M、Hewapathirana S、Kundu DJ、Inuganti A、Griss J、Mayer G、Eisenacher M 等 (2019)2019年的PRIDE数据库及相关工具和资源:改进对量化数据的支持。 核酸研究 47:D442–D450 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
68 Edgar R,Domrachev M,Lash AE(2002)基因表达综合:NCBI基因表达和杂交阵列数据库。 核酸研究 30: 207–210 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
关联数据 本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。