介绍
代谢改变最近已成为癌症的标志之一(1). 癌细胞的复制速度更快,因此具有较高的生物合成和生物能量需求。为了满足这种日益增长的需求,癌细胞的营养吸收和代谢途径发生了改变。即使在氧气充足的情况下,癌细胞也依赖糖酵解而不是氧化磷酸化来获取能量(2). 最近对癌症代谢的研究揭示了替代能源,特别是谷氨酰胺和其他氨基酸在细胞增殖和维持中的作用(三——5)包括癌基因和抑癌基因参与调节癌细胞的代谢途径(6——8).
最近的研究表明c-MYC公司是三阴性乳腺癌(TNBC;参考文献。9). 高c-MYC改变谷氨酰胺分解代谢,显著提高谷氨酰胺吸收,改变谷氨酰胺代谢途径,以维持氧化还原平衡和细胞生长的燃料能量(10)使其成为新的治疗靶点(11). 氨基氧乙酸(AOA)是磷酸吡哆醛依赖酶(包括转氨酶)的一般抑制剂,参与氨基酸代谢,在临床前研究中作为单一药物显示出显著的抗肿瘤作用(10,12,13). 耳鸣患者的临床试验(14,15)和亨廷顿氏病(16),AOA在约1至2 mg/kg/d时耐受性良好。在这些水平下,AOA增加了循环和尿中氨基酸的水平(15,16).
我们推测AOA通过抑制转氨酶耗尽氨基酸库来抑制细胞生长。然后,氨基酸剥夺触发内质网(ER)应激,导致未折叠蛋白反应(UPR;参考文献。17). 内质网应激途径的持续激活将导致细胞凋亡的诱导(18).
在此,我们报告了AOA在免疫缺陷小鼠乳腺癌异种移植模型和免疫效率低下的c-myc转基因小鼠乳腺癌模型中的作用模式和抗肿瘤作用的临床前研究结果。数据支持AOA作为代谢抑制剂的治疗作用,特别是在c-MYC过度表达的乳腺癌中。
翻译相关性。
转氨酶抑制剂氨基氧乙酸(AOA)以谷氨酰胺分解途径为靶点,并在c-MYC临床前模型中显示出强大的抗肿瘤作用,该模型过度表达ER阳性和ER阴性乳腺癌。对AOA的作用机制、有效性和耐受性的了解使其成为进一步临床翻译的最佳候选。
材料和方法
细胞系和试剂
使用的乳腺癌细胞系是从ATCC购买后6个月内冷冻的细胞系(使用STR图谱分析进行鉴定),如下所示:MCF-7(ER/PR+ve(虚拟)/HER2-阴性);SKBR3、HCC1954、HCC202(ER/PR-阴性/HER2+ve(虚拟)); BT474(ER/PR+/HER2型+ve(虚拟)); MDA-MB-231、HCC1806、HCC1143(来自ATCC)、SUM149和SUM159(S.Ethier、MUSC、SC;ER/PR/HER2阴性或三阴性)。这两种细胞系未经独立鉴定。AOA和生化药品从西格玛公司购买。从缩小乳房成形术标本中分离出正常人乳腺上皮细胞(HMEC),并在MCF10A培养基(ATCC)中培养。人类乳房类有机物是通过酶消化还原乳房成形术组织制备的,这些组织是根据IRB批准的方案收集的。从多西环素诱导的MMTV-rTtA-TetO-myc小鼠的原发性乳腺肿瘤中建立小鼠肿瘤细胞系MTC1和MTC2,而MG1和MG2是FVB/n幼崽的原发乳腺。
MTT分析
细胞在100μL培养基中以每孔1500至5000个细胞的速度在96 well板中电镀。12小时后添加不同浓度AOA的新培养基。48小时后进行测定(19).
天冬氨酸转氨酶测定
如前所述,天冬氨酸转氨酶的酶活性通过比色法测定,以评估草酰乙酸(GOT1/2(也称为AST1/2)活性的产物)形成丙酮酸的情况(20). 简言之,在AOA处理6、24或48小时后收集在6孔板中生长的细胞,并用冷PBS洗涤、裂解和用于分析的上清液。
蛋白质印迹分析
使用的抗体如下:抗c-MYC(Abcam)、GRP78、PERK、IRE1a、CHOP、pAMPK、TAMPK、PARP、c-PARP、c-Cas3(细胞信号技术)、cyclin D1、ATF3(Invitrogen)、β-actin(Sigma)。使用ImageJ软件进行定量。
磁共振波谱
AOA处理SUM159细胞24小时。用胰蛋白酶法收集贴壁细胞并计数活细胞。采用双相萃取法获得水溶性和脂质提取物(21).
细胞周期分析和BrdUrd掺入分析
处理后将SUM149或SUM159细胞胰蛋白酶化,并在−20°C的80%乙醇中固定过夜。随后,将细胞重新悬浮在含有20μg/mL碘化丙啶(PI)和10μg/mL核糖核酸酶a的PBS溶液中,并在37°C下培养30分钟(22). 对于溴脱氧尿苷(BrdUrd)掺入,在37°C下用BrdUrd(Invitrogen)处理细胞60分钟,并用PBS洗涤两次(23).
动物实验
所有动物研究均由约翰·霍普金斯大学动物护理和使用委员会批准。将200万SUM149、SUM159、HCC1954、MDA-MB-231或500万MCF-7细胞重新悬浮在50μL PBS和Matrigel(BD Biosciences;1:1)中,皮下注射到运动Balb/c小鼠(NCI、Frederick)。每周测量两次肿瘤。
转基因小鼠模型:如前所述,产生多西环素诱导的乳腺特异性myc过表达小鼠(MMTV rTtA TetO-myc)(24).
统计分析
使用GraphPad Prism(v5.0,GraphPad-Software)和SAS软件(v9.2,SAS Institute)分析定量数据,并在适当情况下表示为平均值±SD或百分比。
每个部分的详细说明见补充方法.
结果
乳腺癌细胞生长对谷氨酰胺的依赖性
乳腺癌细胞对谷氨酰胺的依赖性尚未得到广泛的分类。细胞在补充有含有谷氨酰胺的10%FBS或不含谷氨酰胺的生长培养基(指定为还原谷氨酰胺培养基)中培养。乳腺癌细胞系MDA-MB-231、SUM149、HCC1806、SUM159和MCF-7的生长依赖于谷氨酰胺的补充。相比之下,通过台盼蓝染色评估,BT474、HCC143、HCC1954、HCC202和SKBR3对谷氨酰胺的生长依赖性显著降低(图1A),并通过菌落形成分析证实(补充图S1A). 谷氨酰胺依赖性MDA-MB-231和SUM149细胞系在谷氨酰胺减少的情况下生长6天,凋亡细胞数量显著增加,而在谷氨酰胺依赖型BT474或HCC202细胞系中没有观察到这种变化(补充图S1B).
图1。
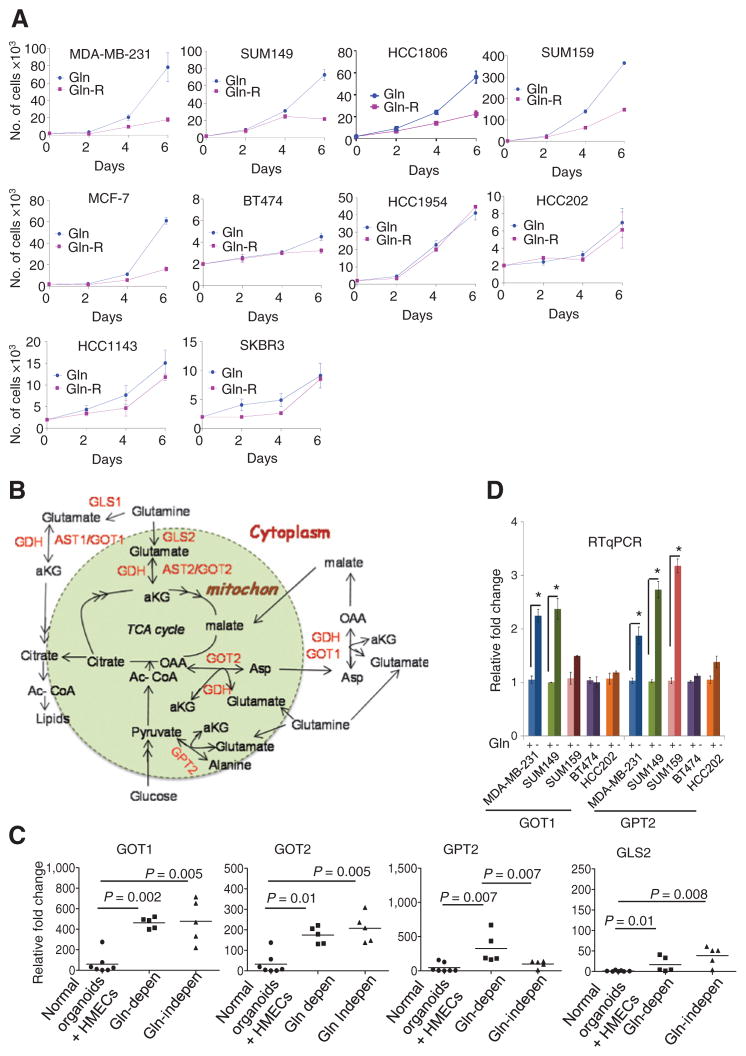
乳腺癌细胞系的谷氨酰胺依赖性。A、 乳腺细胞系在含有完整或还原谷氨酰胺(Gln和Gln-R)的培养基中生长。显示了指定日期每孔(3孔/时间点;两次重复)的平均活细胞数。B、 转氨酶和其他与谷氨酰胺分解有关的基因的示意图。C、 mRNA差异表达GOT1、GOT2、GLS2、和GPT2项目在乳腺癌细胞系中与正常乳腺类器官和HMEC进行比较(每个试验重复;三次重复)。曼惠特尼测试,*,P(P)< 0.05. D、 在具有完全(Gln+)或还原谷氨酰胺(Gln−)24小时(每次试验重复;三次重复);Mann-Whitney检验;*,P(P)< 0.05.
谷氨酸分解途径中基因的差异表达和对AOA的敏感性
谷氨酰胺通过谷氨酸脱氢酶(GDH)或谷氨酸草酰乙酸转氨酶的活性,以α-酮戊二酸的形式进入线粒体,为TCA循环提供碳元素。谷氨酰胺中的氮对核苷酸和氨基酸的生物合成至关重要,谷氨酸和天冬氨酸是合成其他非必需氨基酸的前体(Schema in图1B; 裁判。25).
肿瘤细胞对谷氨酰胺的可变依赖性可能是谷氨酰胺分解途径相关基因表达改变的结果。通过RT-qPCRGOT1、GOT2、GPT2、和GLS2级(图1C),但不是GLS1级和GDH公司(补充图S1C)乳腺癌细胞系中的表达水平明显高于正常上皮类器官和培养的HMEC。有趣的是,GPT2项目与谷氨酰胺依赖性细胞系相比,谷氨酰胺依赖型细胞系的表达显著增高(图1C). 谷氨酰胺停药24小时后获得1在MDA-MB-231和SUM149细胞中显著增加(图1D和补充图S1D)但在谷氨酰胺依赖型BT474和HCC202中保持不变,表明GOT1是谷氨酰胺水解的关键酶之一。GPT2催化可逆的转氨反应,从丙酮酸和谷氨酸中生成α-酮戊二酸和丙氨酸。GPT2型所有三种谷氨酰胺依赖细胞系的mRNA均高于非依赖细胞系(图1D). 这些结果表明,GPT2升高也是一种关键酶,有助于燃料电池在谷氨酰胺缺乏的环境中存活。目前尚无针对GOT或GPT2的特异性抑制剂。因此,我们使用AOA测试了泛转氨酶抑制对10ER存活率的影响+和ER−乳腺癌细胞系在完全培养基中生长。与谷氨酰胺依赖性较低的细胞相比,谷氨酰胺依赖细胞系显示AOA对细胞生长的抑制作用更强(图2A和补充图S2A). 为了确定AOA的靶向酶活性,我们使用耗尽了每种酶的SUM149细胞系(使用siRNA),并测试了它们对AOA的敏感性。如所示图2BGOT1、GOT2或GPT2的缺失部分逆转了AOA介导的细胞毒性作用。当细胞被GOT1+GOT2或GOT1/2+GPT2耗尽时,细胞对AOA的敏感性降低。有趣的是,当细胞GPT2耗尽时,GOT1和GOT2水平都显示出1.6倍的增加(图2C). 这些结果得出结论,GOT1/2和GPT2对AOA介导的效应负有部分责任,也表明了GPT2耗竭后的代偿效应,通过敲低这种酶导致细胞增加其他酶的水平。此外,在SUM149细胞中,这些基因的单独或联合敲除也会影响细胞增殖,这强调了这些基因在细胞生存中的重要性(补充图S2B). AOA也以时间依赖的方式显著抑制了三种细胞系中测试的GOT1/2的酶活性(图2D).
图2。
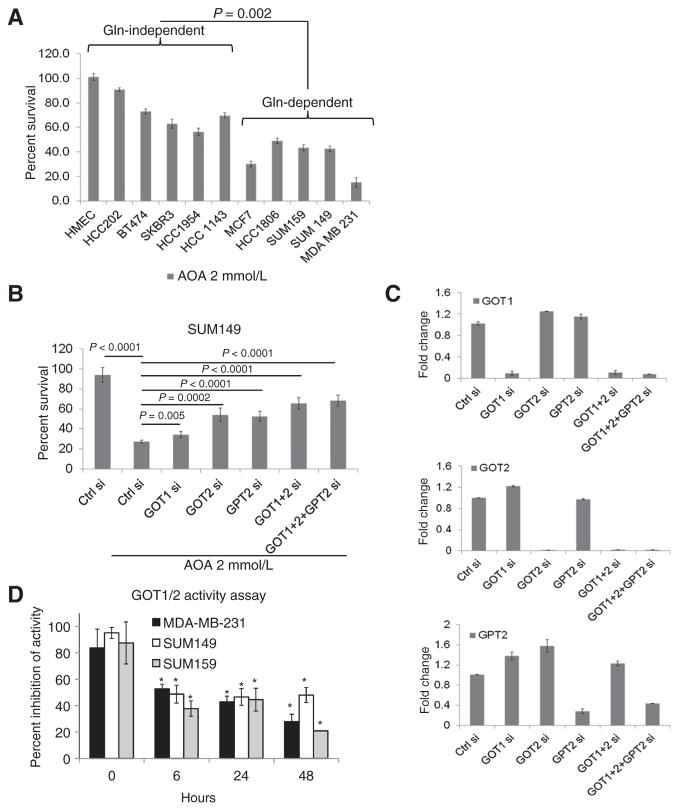
乳腺癌细胞对AOA的敏感性及其在GOT1/2和GPT2缺失的细胞中的逆转。A、 用2mmol/L AOA处理48小时后,对乳腺癌细胞株和正常HMEC进行MTT分析。B、 逆转AOA对GOT1、GOT2和GPT2缺失的SUM149细胞的细胞毒作用。C、 RT-qPCRGOT1、GOT2、和GPT2项目SUM149细胞中的mRNA缺失所示基因。D、 AOA处理(2 mmol/L)后GOT1和GOT2的酶活性在暴露6小时后显示出显著抑制。每个实验重复三次。学生的所有统计分析t吨测试;双尾;*,P(P)< 0.05.
AOA对乳腺癌细胞的作用依赖于c-MYC
在乳腺癌中发现的最早的分子畸变中,据报道,c-MYC扩增和过度表达在30%至50%的乳腺癌中发生(26). 先前已经研究过c-MYC对代谢,特别是谷氨酰胺代谢的整体影响(27,28). c-MYC转录激活谷氨酰胺转运体和谷氨酰胺酶(29). 在胶质母细胞瘤中,c-MYC在谷氨酰胺成瘾中的作用已被充分研究(30)和骨肉瘤(10),但其对乳腺癌代谢的影响尚不明确。一组细胞系的Western blot分析显示,50%的肿瘤细胞中c-MYC水平高于正常水平,c-MYC水平与谷氨酰胺依赖性和AOA敏感性相关,Spearman相关系数r=0.664;P(P)= 0.03 (图3A). 为了测试高c-MYC在AOA作用中的重要性,我们制作了c-MYC缺失的TNBC细胞。SUM149和SUM159中c-MYC的瞬时敲除(图3B)和SUM159细胞的稳定耗竭(图3B)使细胞的敏感性大大降低(P(P)<0.05)对AOA介导的细胞死亡。与对照细胞相比,SUM159-sh-cMYC细胞对谷氨酰胺的依赖性较低(图3B)并且显示谷氨酰胺分解基因的表达显著降低,GOT1公司和GPT2项目,但不是GDH公司或政府2(补充图S3A). 相反,1954年肝癌中c-MYC的过度表达(c-MYC低)增加了AOA敏感性(P(P)< 0.05;图3C). 总之,这些结果表明,在c-MYC高表达的细胞中,AOA敏感性明显更高。这一发现通过使用两种小鼠乳腺肿瘤细胞系MTC1和MTC2进行验证,这两种细胞系是从多西环素诱导的FVB/N小鼠MMTV-rTtA-TetO-myc驱动的乳腺肿瘤中建立的。这两种细胞系均表达高c-myc(维持在含强力霉素的培养基中),并被发现具有谷氨酰胺依赖性(P(P)< 0.001;补充图S3B和S3C)和对AOA的生长抑制敏感(P(P)< 0.05;图3D). 利用诱导型c-myc公司在这些细胞中,我们测试了c-myc表达在其对AOA反应中的重要性。通过从培养基中取出强力霉素,MTC2细胞中c-myc的表达逐渐减少。随着c-myc水平的下降,MTC2细胞在第12天对AOA的敏感性显著下降(补充图S3D)支持c-myc高表达在乳腺癌肿瘤细胞对AOA的反应中发挥重要作用。
图3。
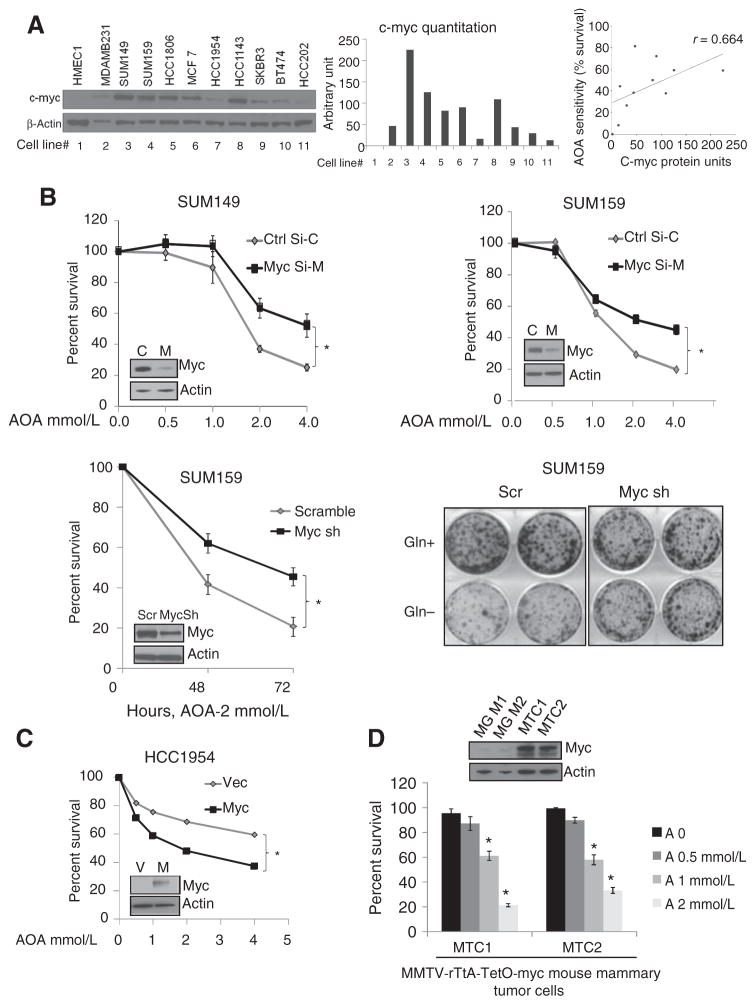
AOA敏感性依赖于c-MYC。A、 乳腺癌细胞c-MYC蛋白的Western blot分析;用负载控制β-actin标准化密度测定评价;以及c-MYC蛋白水平和AOA敏感性之间的相关性。用2mmol/L AOA处理48小时后存活的细胞百分比(来自图2A)绘制同一细胞系中c-MYC蛋白的数量。斯皮尔曼相关第页= 0.664;*,P(P)< 0.05. B、 siRNA介导的SUM149和SUM159细胞c-MYC耗竭。AOA处理48小时的细胞的MTT分析。稳定表达c-MYC shRNA的SUM159细胞与表达scramble(Scr)shRNA的细胞(左)相比对AOA的敏感性;集落形成分析显示,MYC缺失的SUM159细胞不依赖谷氨酰胺生长(右)。C、 MTT法检测AOA对过度表达外源性C-MYC的HCC1954乳腺癌细胞的影响。D、 MTT法检测AOA对两种小鼠乳腺癌细胞株的影响。Western blot分析显示,与未诱导FVB/N小鼠乳腺MG1和MG2相比,c-myc在两种肿瘤细胞系MTC1和MTC2中过度表达。数值表示为三个独立实验的平均值±标准差,在B至D的每个条件下测试4个细胞。所有数据由学生分析t吨测试,双尾;*,P(P)< 0.05.
AOA处理细胞代谢变化的磁共振波谱分析
用磁共振波谱(MRS)定量测量经AOA处理的SUM159细胞中代谢物的变化,补充图S4A; 裁判。31)天冬氨酸和丙氨酸显著降低,谷氨酸含量无明显变化(图4A)表明AOA介导的天冬氨酸和丙氨酸转氨酶抑制。AOA治疗后,我们还观察到与肿瘤转化相关的两种代谢物总胆碱(磷酸胆碱+甘油磷酸胆碱+游离胆碱)和磷酸胆碱的减少(补充图S4B; 裁判。32). 排除AOA对糖酵解途径的影响,在从AOA处理的SUM159细胞收集的条件培养基中测量的乳酸生成和葡萄糖消耗没有明显变化(补充图S4C). 因此,MRS分析进一步证实了AOA在消耗丙氨酸和天冬氨酸方面的作用,并检测到肿瘤形成、总胆碱和磷酸胆碱的既定标记物减少。
图4。
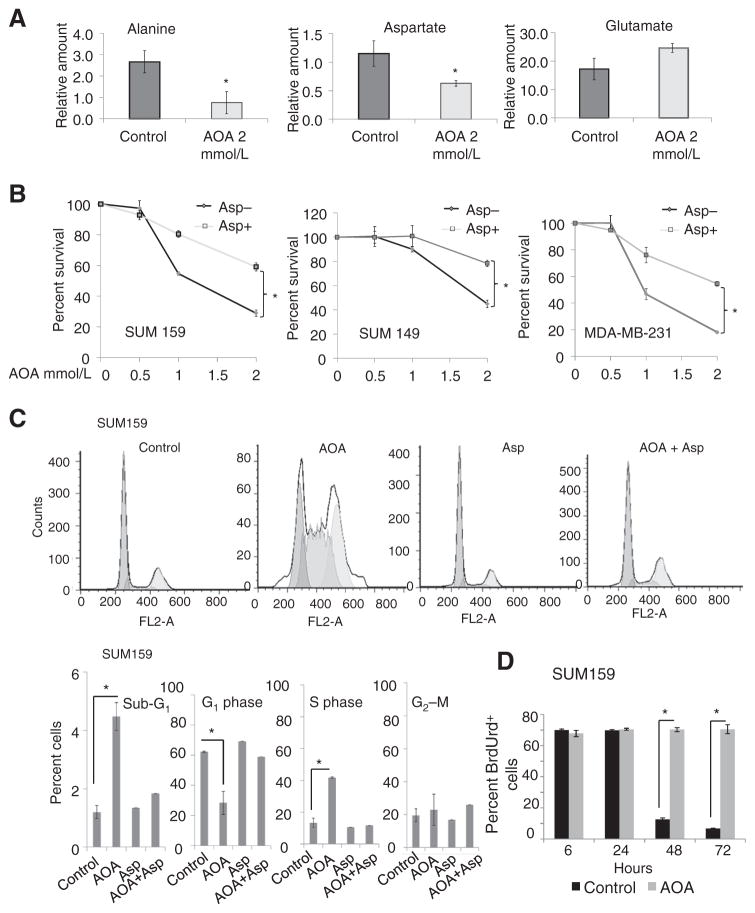
A、 经AOA处理的SUM159细胞的MRS分析显示丙氨酸和天冬氨酸显著减少,但谷氨酸没有减少(*,P(P)< 0.05). B、 MTT分析显示在含有不同量AOA的10 mmol/L天冬氨酸存在下培养48小时的活细胞百分比(*,P(P)< 0.01). C、 AOA诱导S期阻滞,通过添加天冬氨酸逆转。使用PI对暴露于载体、AOA、天冬氨酸或AOA+天冬氨酸酯72小时的SUM159细胞的细胞周期进行流式细胞术分析,显示亚G增加1AOA处理的细胞中的含量和S期阻滞;用天冬氨酸进行共处理可以逆转这种效应。D、 BrdUrd掺入和保留对S期阻滞的稳定性。对细胞进行BrdUrd和PI染色,流式细胞仪分析显示,AOA(2mmol/L)处理48和72小时后,BrdUrd阳性细胞显著增加。数值表示为A到D的三个独立实验的平均值±SD。所有数据均由学生分析t吨测试,双尾。
MRS数据表明,AOA会导致天冬氨酸和丙氨酸的消耗,这两种氨基酸对许多乳腺癌细胞的生长至关重要。如果是这样,外源性添加这些氨基酸可以使细胞免于AOA诱导的细胞死亡。在存在或不存在丙氨酸或天冬氨酸的情况下,用不同剂量的AOA处理培养的MDA-MB-231、SUM149和SUM159细胞。在我们的实验条件下,丙氨酸不能拯救细胞免受AOA诱导的毒性(补充图S4D和S4E)而天冬氨酸使细胞对AOA的敏感性降低(图4B). 这些数据表明,AOA引起的细胞死亡主要是通过天冬氨酸耗竭发生的。
AOA引起S期细胞周期阻滞,天冬氨酸逆转
天冬氨酸是核苷酸生物合成中的氨基供体。进入细胞周期需要最佳的核苷酸水平(33). 因此,AOA对天冬氨酸的消耗可能会影响核苷酸的生物合成,导致细胞周期在S期停滞。指数级增长的SUM159约占13%至15%(图4C)和SUM149电池(补充图S5A)处于S相。AOA治疗后,S期分数增加(分别为32%和38%),G期细胞随之减少1观察到相。外源性天冬氨酸逆转AOA诱导的S期阻滞。子G1AOA处理的细胞中代表凋亡细胞的分数也显著增加(图4C和补充图S5A). 为了评估S期阻滞的稳定性,用BrdUrd对SUM159细胞进行脉冲处理,并用新鲜培养基或2mmol/L AOA处理。随着每一次加倍,对照细胞逐渐失去结合的BrdUrd。然而,在AOA处理的细胞中,BrdUrd-阳性细胞的数量在72小时内保持不变,表明细胞周期持续停滞(图4D). SUM149细胞也获得了类似的结果(补充图S5B). 此外,AOA诱导的细胞周期阻滞是不可逆的,因为持续治疗SUM149和SUM159细胞10天会导致细胞死亡(补充图S5C和S5D). 我们还评估了外源性天冬氨酸能否替代谷氨酰胺并逆转细胞死亡。将谷氨酰胺依赖性MDA-MB-231、SUM149、SUM159和HCC1806细胞系培养在正常培养基、谷氨酰胺还原培养基或谷氨酰胺还原补充外源性天冬氨酸的培养基中。外源性天冬氨酸能够部分挽救这些细胞系中谷氨酰胺缺乏介导的细胞死亡(补充图S5E).
AOA介导细胞凋亡的机制
代谢产物的MRS分析表明,AOA的作用在很大程度上是通过阻断氨基酸代谢来实现的。这反过来又可以诱导细胞中AMPK(活化蛋白激酶)的激活和ER应激。持续的内质网应激通过凋亡导致细胞死亡(18). 为了检测这些途径,我们用AOA处理SUM149和SUM159细胞24小时,并通过RT-qPCR和免疫印迹分析评估mRNA表达和蛋白水平。AOA治疗显著增加了内质网应激标记物的mRNA水平,如ATF3、CHOP,但不是PERK公司(胰腺ER激酶)或ATF6型(图5A). 接下来,我们分析了这些改变是否是AOA敏感细胞系特有的。AOA处理(2mmol/L,72小时)后,内质网应激标记物IRE-1a、GRP78、CHOP和凋亡指示物裂解caspase-3的Western blots分析显示,与对照BT474或HCC202细胞系相比,SUM159细胞中应激标记物的水平显著增加(图5B). GRP78是三个关键传感器ATF6、PERK和IRE-1a的内质网应激调节剂和伴侣,在SUM159细胞中减少了30%。AOA治疗后观察到IRE-1a(与GRP78分离后的ER应激信号转导子)增加3倍,CHOP(ER应激途径中的凋亡诱导剂)增加12倍。在SUM159细胞中检测到裂解的caspase-3增加了15倍。这些数据支持AOA介导的作用在谷氨酰胺依赖性细胞系中的特异性,但在谷氨酰胺诱导依赖性细胞株中没有。此外,细胞周期蛋白D1水平在24小时内下降,AMPK被磷酸化。在SUM159和SUM149细胞中,作用于CHOP上游的ATF3水平升高。PARP裂解,细胞死亡的另一个指标,也被检测到(补充图S6A和S6B). SUM159细胞中添加天冬氨酸部分逆转了AOA诱导的IRE-1a和CHOP增加,GRP78和裂解caspase-3水平下降(图5C).
图5。
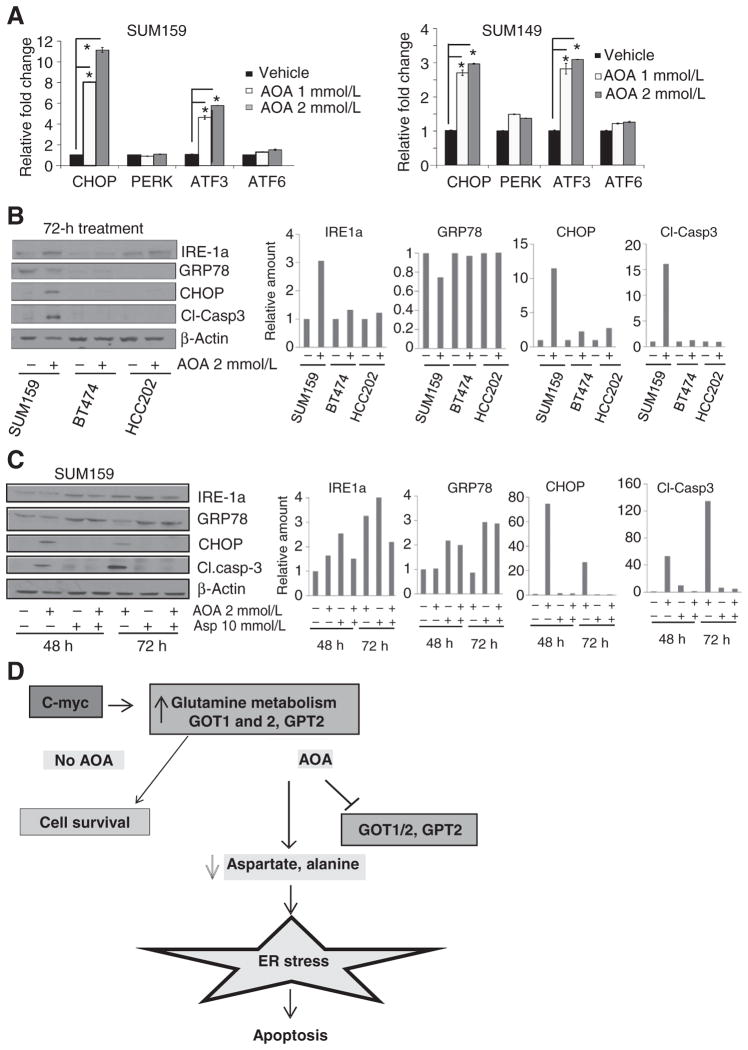
AOA诱导生长抑制和死亡的分子机制。A、 SUM159和SUM149细胞的RT-qPCR分析显示,暴露于2 mmol/L AOA 24小时后,内质网应激标记物被诱导(每次检测重复;三次重复检测)。学生t吨测试,双尾*,P(P)≤ 0.05. B、 在暴露于2 mmol/L AOA 72小时后,对AOA敏感的SUM159和AOA不敏感的BT474和HCC202细胞株进行应激标记物和凋亡标记caspase-3的Western blot分析。C、 AOA处理48或72小时后,通过向SUM159和SUM149细胞添加天冬氨酸来逆转ER应激标记物和caspase-3的诱导;条形图,定量。D、 AOA在乳腺癌细胞中的作用机制。
我们还检测了AOA联合普通化疗药物卡铂治疗后MDA-MB-231细胞中的ER应激标记物。卡铂对应激标记物没有显著影响,但与单独AOA相比,联合用药对mRNA和蛋白质水平的增加具有加性效应。其他标记也显示出类似的模式(补充图S6C和S6D). 总的来说,这些数据为AOA通过GRP78、IRE-1a或CHOP诱导强大的内质网应激反应提供了有力证据,这可能是治疗后观察到的细胞死亡的原因。概述AOA在c-MYC过度表达癌细胞中作用机制的模式如图所示图5D.
AOA在动物模型中的抗肿瘤作用
细胞培养分析表明,AOA具有生长抑制作用,特别是对表达高水平c-MYC蛋白的细胞。我们测试了AOA对三种高c-MYC表达的SUM149、SUM159和MCF-7乳腺癌细胞系和一种低c-MYC-表达细胞系HCC1954作为阴性对照的异种移植瘤的抗肿瘤作用。AOA(5 mg/kg)治疗导致SUM149的肿瘤生长显著降低(P(P)<0.01),总和159(P(P)< 0.001;图6A和补充图S7A和S7B)和MCF-7(P(P)= 0.001;补充图S7C)异种移植物与载体对照相比,但在MYC低表达、阴性对照的HCC1954肿瘤中没有(图6A). AOA治疗的SUM159和SUM149肿瘤的免疫组织化学分析显示,裂解的caspase-3水平较高(补充图S7D). 内质网应激标记物PERK、IRE1和CHOP的蛋白水平在接受AOA治疗的SUM159和SUM149肿瘤中均较高,但在HCC1954异种移植物中不高(图6B和补充图S7E–S7G). 接下来,我们测试了AOA联合卡铂和紫杉醇(临床上常用的治疗TNBC的药物)的疗效。接受AOA治疗的免疫缺陷受体小鼠体重没有下降(补充图S8A). 与单独使用紫杉醇相比,AOA与紫杉醇联合使用显著抑制SUM149肿瘤生长(补充图S8B),但卡铂没有成瘾作用(补充图S8C). 化疗不能增强AOA对SUM159肿瘤的抗肿瘤作用(补充图S8D和S8E).
图6。
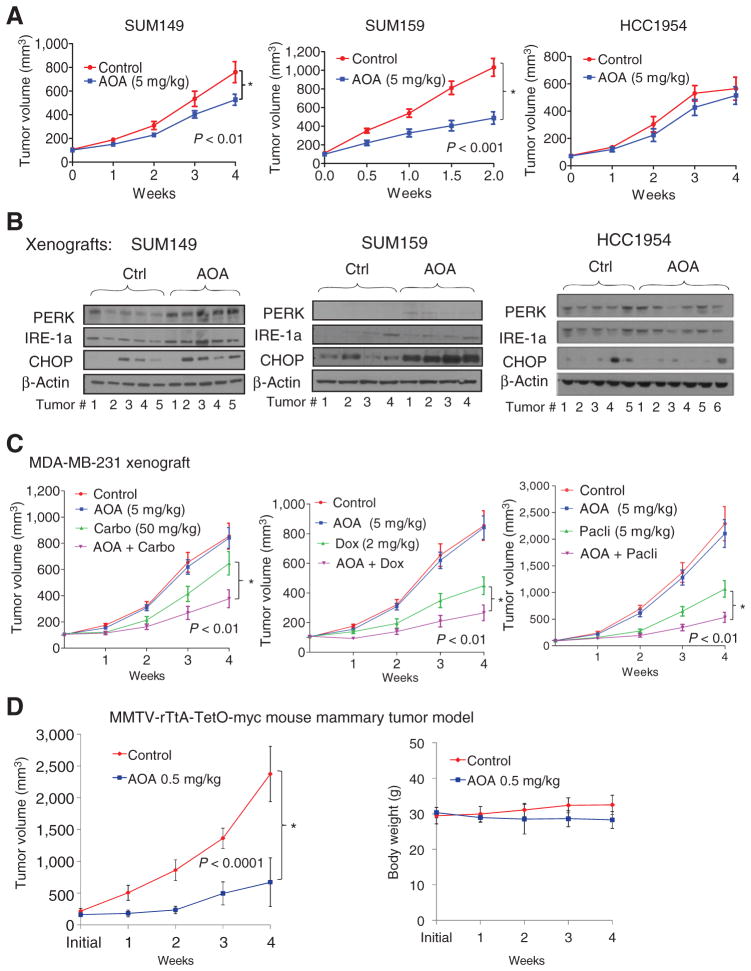
体内AOA的生长抑制作用。A、 将SUM149、SUM159或HCC1954细胞皮下注射到无胸腺Balb/c小鼠中。当肿瘤达到100毫米时三,将小鼠随机分为对照组(PBS)或AOA组(每天5 mg/kg腹腔注射)(n个=6-8只小鼠/组)。B、 SUM159、SUM149和HCC1954异种移植物应激途径标记物的Western blot分析。实验结束时收集肿瘤。C、 在MDA-MB-231异种移植物中,AOA与阿霉素、卡铂或紫杉醇的组合显示出生长抑制作用。数据绘制为平均值±SEM,并通过混合效应模型与Tukey程序进行比较,以进行多次测试修正。D、 左侧,用PBS治疗荷瘤MMTV-rTtA-TetO-myc小鼠(n个=10)或AOA 0.5 mg/kg(静脉注射3天/周;n个= 5). 当肿瘤达到150至200毫米时开始治疗三并持续4周。没错,接受AOA治疗的动物的体重与车辆治疗的动物相比没有显著下降。方差分析;*,P(P)= <0.0001.
在MDA-MB-231细胞中发现了这些观察结果的例外。在这里,尽管c-MYC的表达处于中等水平,但在培养中,细胞对AOA非常敏感。此外,在单独用AOA处理的MDA-MB-231细胞中,ER应激和凋亡途径发生激活,当AOA与卡铂联合使用时,效果更为明显(补充图S6C和S6D). 将AOA作为单一试剂处理后,MDA-MB-231异种移植物未表现出任何生长抑制(图6C). 在这里,AOA与阿霉素、卡铂或紫杉醇的联合使用导致显著的(P(P)<0.01)肿瘤缩小。对一组代表性肿瘤进行的Western blot分析表明,与载体对照或AOA治疗相比,卡铂或阿霉素单独治疗导致c-MYC蛋白水平增加近2倍(补充图S8F). 这些数据增加了AOA敏感性不仅与成分相关,而且与化疗诱导的c-MYC蛋白过度表达相关的可能性。此外,AOA作为单一药物或与化疗联合治疗的有效性可以通过预处理肿瘤活检中的c-MYC水平来预测。此外,c-MYC治疗后上调可能有助于提高AOA的敏感性。
为了进一步证实我们的发现,我们使用了多西环素诱导MMTV驱动的myc过度表达转基因小鼠模型。强力霉素诱导后,双转基因小鼠发生乳腺肿瘤的潜伏期约为22周(24). 当肿瘤大小达到150至200毫米时三,小鼠每周3天以0.5 mg/kg体重的AOA治疗4周(n个=5–10/组)。AOA治疗小鼠的肿瘤生长显著降低(图6D). 与Balb/c nu/nu小鼠相比,AOA剂量超过0.5 mg/kg(测试剂量为0.5、1、2.5和5 mg/kg)会导致该种小鼠体重减轻和死亡。
讨论
谷氨酰胺代谢对乳腺癌增殖的作用尚未得到广泛研究,尤其是在TNBC中。在这项研究中,我们提供了证据表明,表达高水平c-MYC的乳腺癌细胞系依赖谷氨酰胺维持生存和生长。使用转氨酶抑制剂AOA抑制这些细胞系中的谷氨酰胺分解,主要通过激活内质网应激途径导致细胞死亡。这些发现使我们进一步将AOA作为乳腺癌的治疗靶点。
抑制谷氨酰胺代谢是一个活跃的研究领域。因此,针对谷氨酰胺代谢的几种药物正在开发中(34). 2008年,研究人员报告称,用AOA处理单个细胞系MDA-MB-231后,丙氨酸和13C-葡萄糖衍生碳转化为谷氨酸和尿苷,并降低耗氧率、细胞ATP水平和NAD+/NADH比率(13). 在这项研究中,我们报道了AOA介导的乳腺癌细胞死亡途径的深入表征。与正常乳腺细胞相比,乳腺癌细胞系中谷氨酰胺代谢所必需的许多酶(包括GOT1、GOT2、GPT2和GLS2)的表达水平和活性都高。这种对替代能源的依赖很可能是乳腺癌细胞对谷氨酰胺上瘾的基础。导致谷氨酰胺成瘾的第二个因素可能是细胞中c-MYC的高水平表达(30). 如果是这样,那么c-MYC过度表达的细胞对转氨酶抑制剂(如AOA)尤其敏感,这是理所当然的。事实上,我们发现c-MYC水平与药物敏感性之间存在显著相关性(图3A-C). 我们的数据与已发表的研究结果一致,在c-MYC过度表达的神经母细胞瘤中,ATF4及其下游调节因子被发现是谷氨酰胺停药导致细胞凋亡的关键介质(12). 在TNBC细胞中,AOA处理降低了伴侣蛋白GRP78的水平,并激活了许多内质网应激途径基因,包括ATF4下游的ATF3。肿瘤微环境中的应激,如低氧、低血糖和氨基酸可用性降低,激活UPR,这是一种由内质网腔内过量错误折叠或未折叠蛋白质触发的细胞内稳态程序(18). 在长期压力下,UPR启动一个导致细胞凋亡的程序。UPR的三个近端效应器是PERK、激活转录因子6(ATF6)和需要肌醇的跨膜激酶/内切酶1(IRE1)。PERK的自身磷酸化可以翻译特定的帽非依赖性内质网应激反应基因,如ATF4。促凋亡蛋白CHOP(CCAAT/-增强子结合蛋白同源蛋白)在ATF4下游上调,导致抗凋亡线粒体蛋白Bcl-2下调,促进凋亡。其次,ATF6在易位到高尔基体后通过蛋白水解裂解被激活(35). IRE1a激活JNK信号通路,在这一点上,人类前caspase-4被认为通过激活下游caspase引起凋亡(18). 哺乳动物腺苷一磷酸活化蛋白激酶(AMPK)是一种丝氨酸-苏氨酸激酶蛋白复合物,是细胞能量平衡的中央调节器;AMPK介导细胞对代谢应激反应的机制尚不清楚(36). 我们在此表明,内质网应激可能是AOA治疗引起的细胞生长抑制的主要途径。
我们还提出了AOA抑制细胞生长的新机制。众所周知,谷氨酰胺和天冬氨酸提供了对核苷合成至关重要的胺基。如我们的细胞周期分析和BrdUrd掺入分析所示,细胞中核苷池的减少可能导致S期细胞周期阻滞(37). 在我们的研究中,单用天冬氨酸能有效降低AOA对乳腺癌细胞的敏感性并逆转S期阻滞,这可能归因于天冬氨酸能挽救核苷酸合成。另一个有趣的发现是在MRS研究中观察到AOA对胆碱代谢的影响。据报道,恶性乳腺肿瘤中磷酸胆碱和总胆碱水平升高(32,38,39). AOA在降低总胆碱和磷酸胆碱中的作用尚待研究。
虽然MDA-MB-231细胞对AOA敏感在体外与公布的报告相反(13)仅观察AOA处理小鼠(10 mg/kg体重)2周,我们没有观察到AOA 5 mg/kg体重时的肿瘤生长抑制。在我们看来,更大剂量与一周后的体重减轻和毒性迹象有关,尤其是与化疗药物联合使用时更为严重。MDA-MB-231细胞是否克服AOA诱导的内质网应激介导的细胞死亡体内通过与微环境的对话需要进一步调查。
AOA对MMTV-rTtA-TetO-myc转基因小鼠乳腺肿瘤有较强的生长抑制作用。通过利用该系统中该基因的多西环素诱导性,我们证明了myc缺失可逆转肿瘤细胞对AOA的敏感性。此外,这些发现为探索这种小分子在c-MYC过度表达乳腺肿瘤中的作用提供了有力的依据。AOA可能是一种极具吸引力的治疗药物,因为它具有高度的耐受性和强大的抗肿瘤作用,为进一步开发药物提供了强有力的理由。
总之,我们已经表明,乳腺癌细胞,尤其是TNBC的生长依赖谷氨酰胺,AOA可以有效地针对这种依赖性。AOA诱导的内质网应激途径导致细胞生长抑制和凋亡。AOA虽然对肿瘤细胞具有细胞毒性,但在小型临床试验中显示出可接受的毒性特征,这可能有助于快速开发药物。此外,由于临床前数据表明谷氨酰胺代谢在多种其他癌症类型中发挥作用,特别是肉瘤、脑肿瘤和其他c-MYC驱动的癌症,AOA可能具有更广泛的治疗应用。
致谢
作者感谢宾夕法尼亚大学Lewis Chodosh博士慷慨提供MMTV-rTtA-etO-myc转基因小鼠,以及A.Goga博士提供c-myc过表达载体。作者感谢Dileep Unnikrishnan和Sidra Hafeez的实验室协助。
赠款支持
本研究得到了国防部卓越中心拨款W81XWH-04-1-0595(给S.Sukumar)的支持;苏珊·科曼治愈博士后基金会(PDF12231403;给P.Korangath);Cindy Rosencrans三阴性乳腺癌研究基金(给V.Stearns)和SKCCC核心赠款P30 CA006973(给S.Sukumar)。
脚注
潜在利益冲突的披露
V.Stearns报告称,他们获得了Abbvie、Abraxis、Medimmune、Merck、Novartis和Pfizer的商业研究资助。其他作者没有透露任何潜在的利益冲突。
作者的贡献
概念和设计:P.Korangath、L.Han、C.V.Dang、V.Stearns、S.Sukumar
方法开发:P.Korangath、N.Mori、C.V.Dang、S.Sukumar
数据采集(提供的动物、采集和管理的患者、提供的设施等):P.Korangath、H.Sadik、N.Mori、F.Wildes、S.Sukumar
数据分析和解释(例如统计分析、生物统计学、计算分析):P.Korangath、W.W.Teo、H.Sadik、N.Mori、S.Bharti、Z.Zhang、H.Tsai、C.V.Dang、V.Stearns、S.Sukumar
撰写、审查和/或修订手稿:P.Korangath、N.Mori、C.M.Huijts、Z.Zhang、C.A.Santa Maria、H.Tsai、V.Stearns、Z.M.Bhujwalla、S.Sukumar
行政、技术或物质支持(即报告或组织数据、构建数据库):P.Korangath、C.M.Huijts、Z.M.Bhujwalla、S.Sukumar
研究监督:C.V.Dang和S.Sukumar
工具书类
-
1Hanahan D,Weinberg RA。癌症的标志:下一代。单元格。2011;144:646–74。doi:10.1016/j.cell.2011.02.013。[DOI程序] [公共医学] [谷歌学者]
-
2Warburg O。关于癌细胞的起源。科学。1956;123:309–14. doi:10.1126/science.123.3191.309。[DOI程序] [公共医学] [谷歌学者]
-
三。DeBerardinis RJ、Lum JJ、Hatzivassiliou G、Thompson CB。癌症生物学:代谢重组刺激细胞生长和增殖。单元格元数据。2008;7:11–20. doi:10.1016/j.cmet.2007.10.002。[DOI程序] [公共医学] [谷歌学者]
-
4Sheen JH,Zoncu R,Kim D,Sabatini DM。亮氨酸剥夺后自噬的缺陷调节揭示了人体黑色素瘤细胞在体内外的靶向性。癌细胞。2011;19:613–28. doi:10.1016/j.ccr.2011.03.012。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
5Possemato R、Marks KM、Shaul YD、Pacold ME、Kim D、Birsoi K等。功能基因组学揭示丝氨酸合成途径在乳腺癌中至关重要。自然。2011;476:346–50. doi:10.1038/nature10350。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
6Gao P、Tchernyshyov I、Chang TC、Lee YS、Kita K、Ochi T等。miR-23a/b的c-Myc抑制增强线粒体谷氨酰胺酶表达和谷氨酰胺代谢。自然。2009;458:762–5. doi:10.1038/nature07823。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
7Maddocks OD、Berkers CR、Mason SM、Zheng L、Blyth K、Gottlieb E等。丝氨酸饥饿诱导癌细胞应激和p53依赖性代谢重塑。自然。2013;493:542–6. doi:10.1038/nature11743。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
8Mauro C、Leow SC、Anso E、Rocha S、Thotakura AK、Tornatore L等。NF-κB通过上调线粒体呼吸来控制能量稳态和代谢适应。自然细胞生物学。2011;13:1272–9. doi:10.1038/ncb2324。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
9Alles MC、Gardiner-Garden M、Nott DJ、Wang Y、Foekens JA、Sutherland RL等。与er状态相关的Meta分析和基因集富集显示,“基础”乳腺癌亚组中MYC和E2F活性升高。《公共科学图书馆·综合》。2009;4:e4710。doi:10.1371/journal.pone.0004710。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
10Anso E、Mullen AR、Felsher DW、Mates JM、Debrardinis RJ、Chandel NS。MYC抑制后癌细胞的代谢变化。癌症转移。2013;1:7. doi:10.1186/2049-3002-1-7。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
11Dang CV.Myc重组癌细胞代谢的治疗靶向性。冷泉Harb Symb Quant生物。2011;76:369–74. doi:10.1101/sqb.2011.76.011296。[DOI程序] [公共医学] [谷歌学者]
-
12Qing G,Li B,Vu A,Skuli N,Walton ZE,Liu X,等。ATF4在谷氨酰胺缺乏时调节MYC介导的神经母细胞瘤细胞死亡。癌细胞。2012;22:631–44. doi:10.1016/j.ccr.2012.09.021。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
13Thornburg JM、Nelson KK、Clem BF、Lane AN、Arumugam S、Simmons A等。靶向乳腺癌中的天冬氨酸转氨酶。2008年乳腺癌研究;10:R84。doi:10.1186/bcr2154。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
14Reed HT、Meltzer J、Crews P、Norris CH、Quine DB、Guth PS。氨基氧乙酸作为耳鸣缓解剂。耳鼻咽喉弓。1985;111:803–5. doi:10.1001/archotol.1985.00800140047008。[DOI程序] [公共医学] [谷歌学者]
-
15Guth PS、Risey J、Briner W、Blair P、Reed HT、Bryant G等。氨基氧乙酸作为耳鸣缓解剂的评估。Ann Otol犀牛喉咙。1990;99:74–9. doi:10.1177/00034894900113。[DOI程序] [公共医学] [谷歌学者]
-
16Perry TL、Wright JM、Hansen S、Allan BM、Baird PA、MacLeod PM。氨氧乙酸治疗亨廷顿病的失败。神经病学。1980;30:772–5. doi:10.1212/wnl.30.7.772。[DOI程序] [公共医学] [谷歌学者]
-
17Hetz C、Chevet E、Harding HP。针对疾病中未展开的蛋白质反应。Nat Rev药物发现。2013;12:703–19. doi:10.1038/nrd3976。[DOI程序] [公共医学] [谷歌学者]
-
18Szegezdi E,Logue SE,Gorman AM,Samali A.内质网应激诱导凋亡的介体。EMBO代表2006;7:880–5. doi:10.1038/sj.embor.70400779。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
19van de Loosdrecht AA、Beelen RH、Ossenkoppele GJ、Broekhoven MG、Langenhuijsen MM。一种基于四氮唑的比色MTT分析法,用于定量人类单核细胞介导的对来自细胞系和急性髓系白血病患者的白血病细胞的细胞毒性。免疫学方法杂志。1994;174:311–20. doi:10.1016/0022-1759(94)90034-5。[DOI程序] [公共医学] [谷歌学者]
-
20Bergmeyer HUBE公司。Reitman和Frankel的比色分析。纽约:学术出版社;1974[谷歌学者]
-
21Glunde K,Raman V,Mori N,Bhujwalla ZM。乳腺癌细胞中RNA干扰介导的胆碱激酶抑制诱导分化和减少增殖。2005年癌症研究;65:11034–43. doi:10.1158/0008-5472.CAN-05-1807。[DOI程序] [公共医学] [谷歌学者]
-
22van Engeland M,Ramaekers FC,Schutte B,Reutelingsperger CP。一种测量培养中粘附细胞凋亡过程中质膜不对称性损失的新方法。细胞测定。1996;24:131–9. doi:10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2米。[DOI程序] [公共医学] [谷歌学者]
-
23Campana D,Coustan-Smith E,Janossy G.研究人类B和T淋巴细胞增殖活性的双重和三重染色方法。免疫学方法杂志。1988;107:79-88。doi:10.1016/0022-1759(88)90012-9。[DOI程序] [公共医学] [谷歌学者]
-
24D’Cruz CM、Gunther EJ、Boxer RB、Hartman JL、Sintasath L、Moody SE等。c-MYC通过涉及自发Kras2突变的首选途径诱导乳腺肿瘤发生。《国家医学》,2001年;7:235–9. doi:10.1038/84691。[DOI程序] [公共医学] [谷歌学者]
-
25Vander Heiden MG、Cantley LC、Thompson CB。了解War-burg效应:细胞增殖的代谢需求。科学。2009;324:1029–33. doi:10.1126/science.1160809。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
26Horiuchi D、Kusdra L、Huskey NE、Chandriani S、Lenburg ME、Gonzalez-Angulo AM等。三阴性乳腺癌中的MYC途径激活与CDK抑制是合成致死的。《实验医学杂志》2012;209:679–96. doi:10.1084/jem.20111512。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
27Liu W,Le A,Hancock C,Lane AN,Dang CV,Fan TW,等。脯氨酸和谷氨酰胺代谢的重新编程有助于肿瘤转录因子C-MYC调节的增殖和代谢反应。美国国家科学院院刊2012;109:8983–8. doi:10.1073/pnas.1203244109。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
28Dang CV.用Myc微观管理谷氨酰胺代谢重新思考Warburg效应。2010年癌症研究;70:859–62. doi:10.1158/0008-5472.CAN-09-3556。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
29Li F,Wang Y,Zeller KI,Potter JJ,Wonsey DR,O'Donnell KA等。Myc刺激核酸编码的线粒体基因和线粒体生物发生。分子细胞生物学。2005;25:6225–34. doi:10.1128/MCB.25.14.6225-6234.2005。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
30Wise DR、DeBerardinis RJ、Mancuso A、Sayed N、Zhang XY、Pfeiffer HK等。Myc调节刺激线粒体谷氨酰胺分解并导致谷氨酰胺成瘾的转录程序。美国国家科学院院刊2008;105:18782–7. doi:10.1073/pnas.0810199105。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
31Kinoshita Y,Yokota A.使用体外质子磁共振波谱测定人脑肿瘤中代谢物的绝对浓度。核磁共振生物识别。1997;10:2–12. doi:10.1002/(sici)1099-1492(199701)10:1<2::aid-nbm442>3.0.co;2个。[DOI程序] [公共医学] [谷歌学者]
-
32Glunde K,Jie C,Bhujwalla ZM。乳腺癌胆碱磷脂代谢异常的分子原因。癌症研究2004;64:4270–6. doi:10.1158/0008-5472.CAN-03-3829。[DOI程序] [公共医学] [谷歌学者]
-
33Ewald B,Sampath D,Plunkett W.核苷类似物:细胞死亡信号的分子机制。致癌物。2008;27:6522-37。doi:10.1038/onc.2008.316。[DOI程序] [公共医学] [谷歌学者]
-
34Bobrovnikova-Marjon E,Hurov JB。针对癌症中的代谢变化:新的治疗方法。2014年医学年鉴;65分157秒至70秒。doi:10.1146/annurev-med-092012-112344。[DOI程序] [公共医学] [谷歌学者]
-
35Crespo I、San-Miguel B、Prause C、Marroni N、Cuevas MJ、Gonzalez-Galego J等。谷氨酰胺治疗可减轻TNBS诱导的结肠炎中的内质网应激和细胞凋亡。《公共科学图书馆·综合》。2012;7:e50407。doi:10.1371/journal.pone.0050407。[DOI程序] [PMC免费文章] [公共医学] [谷歌学者]
-
36Wirth M、Joachim J、Tooze SA。自噬体形成——ULK1和Beclin1-PI3KC3复合物在设置阶段中的作用。塞明癌症生物学。2013;23:301–9. doi:10.1016/j.semcancer.2013.05.007。[DOI程序] [公共医学] [谷歌学者]
-
37Jordheim LP、Durantel D、Zoulim F、Dumontet C。用于癌症和病毒疾病的核苷和核苷酸类似物的开发进展。Nat Rev药物发现。2013;12:447–64. doi:10.1038/nrd4010。[DOI程序] [公共医学] [谷歌学者]
-
38Jacobs MA、Barker PB、Bottomley PA、Bhujwalla Z、Bluemke DA。人类乳腺癌的质子磁共振波谱成像:初步研究。J Magn Reson成像。2004;19:68–75. doi:10.1002/jmri.10427。[DOI程序] [公共医学] [谷歌学者]
-
39Baik HM,Su MY,Yu H,Mehta R,Nalcioglu O。使用水作为内部参考物,通过1H核磁共振波谱法对恶性乳腺肿瘤中的胆碱化合物进行定量。5吨岩浆。2006;19:96–104. doi:10.1007/s10334-006-0032-4。[DOI程序] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。

