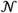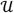介绍 定义染色体拷贝数和等位基因比率是理解癌症基因组结构和历史的基础。 当前的基因组表征技术以基因组单位(DNA质量)测量癌症样本中的体细胞变化。 这种测量的意义取决于肿瘤的纯度及其整体倍性; 因此,它们很难在样本之间进行解释和比较。 理想情况下,拷贝数应该以癌细胞拷贝数来衡量。 这样的测量结果很容易解释,对于癌细胞群中固定的变化来说,是简单的整数值。 这比测量肿瘤衍生样本中二倍体DNA质量单位的相对拷贝数更具挑战性。
使用微阵列在相对基础上测量体细胞拷贝数变化(SCNA)是很简单的 1 , 2 , 三 , 4 , 5 或大规模并行测序技术 6 , 7 ; 自比较基因组杂交(CGH)发展以来,它一直是拷贝数分析的标准方法 8 .
推断绝对拷贝数更加困难,因为:(i)癌细胞几乎总是与未知比例的正常细胞混合(肿瘤纯度); (ii)癌细胞的实际DNA含量(倍性)未知,这是由染色体的数量和结构异常引起的 9 , 10 , 11 , 12 , 13 (iii)癌细胞群体可能是异质的,可能是由于正在进行的亚克隆进化 14 , 15 原则上,可以根据每个癌细胞DNA质量的细胞学测量结果,通过重新调整相关数据来推断绝对拷贝数 16 , 17 , 18 ,或通过单细胞测序方法 15 然而,这种方法不适合支持癌症基因组综合表征的初始大规模工作 19 .
我们几年前开始关注这个问题,最初发展 特别的 技术 20 , 21 随后,我们开发了一种完全定量方法(ABSOLUTE),并将其应用于多个癌症基因组分析项目,包括癌症基因组图谱(TCGA)项目。 ABSOLUTE为在绝对(细胞)基础上对癌症基因组改变进行综合基因组分析奠定了基础。 我们使用这些方法将纯度和倍性估计值与表达亚型相关联,并开发统计能力计算,并使用它们为几个已发表的全基因组测序选择性能良好的样本 22 , 23 , 24 以及许多正在进行的项目,包括乳腺癌、前列腺癌和皮肤癌基因组特征分析。 最近,我们扩展了ABSOLUTE,以推断每个癌细胞在整数等位基因单位中的体细胞点突变的多样性。
我们在这里的目的是:(i)介绍ABSOLUTE方法的数学推理框架,以及其预测的实验验证; (ii)将其应用于分析大型癌症数据集,从而能够对肿瘤演化过程中全基因组加倍的发生率和时间进行新的表征; 描述一种新的点突变和拷贝数估计的综合分析及其在卵巢癌中的应用。
我们描述了ABSOLUTE的三个关键数学特性。 首先,它直接从观察到的相对拷贝图谱(如果可用,也可以使用点突变)联合估计肿瘤纯度和倍性。 其次,由于联合估计可能无法在单个样本上完全确定,因此它使用大量不同的样本集合来帮助解决不明确的情况。 第三,该模型试图解释亚克隆拷贝改变和点突变,这是异质性癌症样本中预期的。
然后,我们报告了第一次大规模的绝对“泛癌”分析,分析了3155个癌症样本的拷贝数变化,代表25种疾病,至少有20个样本。 分析表明,全基因组加倍事件在肿瘤发生过程中频繁发生,最终导致由加倍细胞衍生的成熟癌症,具有复杂的核型。 尽管有证据表明基因组加倍会导致遗传不稳定并加速癌症的发生 25 , 13 , 26 此类事件的发生率和发生时间在人类癌症中还没有广泛的特征。
然后,我们描述了肿瘤纯度和绝对拷贝数的估计如何使我们能够分析测序数据,以区分克隆和亚克隆点突变,并检测卵巢癌样本中的宏观亚克隆结构。 克隆事件在癌细胞中可分为纯合性或杂合性,指导对其功能的解释。 此外,量化点突变的整数多重性的能力将DNA获得之前发生的事件(包括突变位点)与之后发生的事件区分开来。
最后,我们的数据可以描述与基因组加倍有关的体细胞癌进化特征,我们在卵巢癌中证明了这一点,并与临床病理参数相关联。
结果 癌源DNA样品纯度和倍性的推断 ABSOLUTE的概念概述如所示 图1 当从癌症和正常细胞混合群体中提取DNA时,绝对拷贝数信息 每个癌细胞 在混合过程中丢失。 ABSOLUTE的目的是从混合DNA群体中重新提取这些数据。 该过程首先生成分段拷贝数数据,并将其输入到ABSOLUTE算法中,再加上复发性癌症核型的预先计算模型,以及体细胞点突变的等位基因分数值(可选)。 然后,ABSOLUTE的输出提供了关于局部DNA片段的绝对细胞拷贝数的重新提取信息,对于点突变,还提供了突变等位基因的数量( 图1 ).
图1。 使用ABSOLUTE进行肿瘤DNA分析概述。
从由癌症和正常细胞组成的异质细胞群中提取出恒定质量的DNA。 使用微阵列或大规模并行测序技术对该DNA进行分析,给出DNA浓度的全基因组剖面图(蓝线)。 ABSOLUTE使用复发性癌症核型的统计模型将DNA浓度解释为离散拷贝状态,对应于主要为克隆的体细胞拷贝数变化,尽管通常存在一些亚克隆变化。 如果体细胞点突变数据可用(来自DNA测序),那么这些突变的等位基因部分(测序读数中含有非参考等位基因的部分)可能有助于解释DNA浓度。 此外,等位基因部分可以被重新解释为每个癌细胞的整数等位基因拷贝(多重性),这可能揭示亚克隆点突变。
我们首先描述ABSOLUTE方法中使用的推理框架。 假设一个癌组织样本由一定比例的混合物组成 α 癌细胞(假定为单基因组-即癌细胞中含有同质SCNA)和比例(1- α )污染正常(二倍体)细胞。 对于基因组中的每个基因座x,让q(x)表示癌细胞中该基因座的整数拷贝数。 让 τ 表示癌细胞部分的平均倍性,定义为基因组中q(x)的平均值。 在混合癌症样本中,基因座x的平均绝对拷贝数为 α q(x)+2(1- α )平均倍性为D= ατ + 2(1 - α ),以单倍体基因组单位计量。
因此,位点x的相对拷贝数为:
因为q(x)取整数值,R(x)则取离散值。 最小可能值为[2(1- α )/D] 发生在纯合缺失位点,对应于正常细胞DNA的部分。 值之间的间距[ α/ D] 对应于每个癌细胞一个拷贝和每个正常细胞0个拷贝的等位基因的浓度比。 重要的是,如果一个癌症样本不是严格克隆的,那么在大量亚克隆片段中发生的拷贝数变化将显示为该模式的异常值( 图1 , 补充图1a-c ,箭头)。
类似的考虑为利用SNP微阵列衍生的等位基因复制比推断纯度和倍性的算法奠定了基础 27 - 31 , 32 我们将绝对拷贝推断扩展到包括体细胞点突变,如下所示:
这里是 q个 表示点突变的多重性,以每个癌细胞的整数值表示(不能超过q(x)),以及D 秒 = αq(x) + 2(1 - α ). F(x)的值对应于支持突变的预期测序读数部分,这取决于样本纯度和突变位点q(x)处的绝对体细胞拷贝数。
ABSOLUTE算法通过联合优化两个参数来检查相对于整数拷贝数的可能映射 α 和 τ ( 补充图1c-d,h-i ; 联机方法 等式5 ). 在许多情况下,可能会有多个这样的映射,对应于多个optima。
为了帮助解决不明确的病例,我们使用了基于大量样本数据集的复发性癌症核型模型( 补充图2 ; 联机方法 等式8 )确定能充分解释数据的最简单(即最常见)的核型。 这种方法倾向于更简单的解决方案,同时保留了灵活性,以便在提供充分证据的情况下,通过拟合复制文件来识别意外的核型。 事实上,使用ABSOLUTE鉴定出了几个不寻常的核型,包括近单倍体(<1.2n)和超非整倍体(>6n)基因组( 补充图2 ).
我们的实现支持从总或等位基因拷贝比率数据进行拷贝数推断,因此可以使用阵列CGH、SNP微阵列或大规模并行测序数据。 ABSOLUTE可从以下网址下载: http://broadinstitute.org/software/ABSOLUTE .
验证 我们使用以下几种方法验证了ABSOLUTE对Affymetrix SNP微阵列数据的纯度和倍性预测:(i)通过荧光激活细胞分选直接测量37个TCGA卵巢癌样本的倍性 33 ( 图2a ); (ii)基于光谱核型分析的33个NCI60细胞系的倍性测量 34 ( 图2b、c ); 和(iii)DNA混合实验,其中癌细胞系与成对的正常B淋巴细胞衍生的DNA以不同的质量比例混合( 图2d ,在线方法)。 我们还评估了一种相关的计算方法ASCAT 30 ,基于这些数据( 图2a-d , 补充说明1 ). 虽然结果与我们的估计大致一致,但ABSOLUTE的结果要准确得多( 图2a-d )我们的验证数据。 值得注意的是,我们观察到ASCAT明显低估了癌细胞分数( 图2 c,d ),与之前在基于Illumina SNP阵列的类似混合实验中应用ASCAT的报告一致 30 , ( 图S4 )表明偏差与测量平台无关。
图2。 ABSOLUTE方法验证和比较a-d ABSOLUTE和ASCAT在4次验证分析中的表现。
均方根误差:均方根误差。 对 -使用配对单侧Wilcoxon检验(*: 对 < 0.05, **: 对 < 0.001). 请参见 补充说明1 用于ASCAT2.1协议。
a、, 37例原发性肿瘤样本基于FACS的倍性测量与推断的倍性估计。 虚线表示 年 = x个 .
b、, 基于SKY的33种癌细胞系倍性测量与推断的倍性估计。 数据显示如下 一 .
c、, 所示33个细胞系的估计纯度( b条 )虚线水平线表示真实性(1.0)。
d、 两种细胞系的癌-正常DNA混合实验结果。 每个癌细胞系的DNA以不同比例与匹配的B淋巴细胞的DNA混合( x个 -轴)。 (顶部)预测与真实DNA混合分数与 年 = x个 线(虚线)。 (底部)预测的癌症细胞系倍性与混合物纯度。 几个样本的复制文件被曲解了(x); RMSE计算中没有包括这些点。 倍性估计值与之前SKY对这些细胞系的分析基本一致: 网址:http://www.path.cam.ac.uk/ ~木鱼/细胞%20line%20cataloges/breast-cell-lines.htm .
e、 组织学肿瘤中白细胞甲基化特征富集低估了纯度。 HGS-OvCa样本根据所示的组织学纯度评估进行分组( x个 -轴) 33 黑色水平线表示各组的中位纯度,由ABSOLUTE估算( 年 -轴)。 每个点的颜色对应于样本甲基化特征与纯化白细胞相似的程度(在线方法)。
值得注意的是,ABSOLUTE对大块肿瘤的纯度估计似乎比冷冻肿瘤切片的组织学检查结果更准确(在线方法, 图2e ). 458例卵巢癌标本中正常细胞污染比例的估计 33 由ABSOLUTE产生的基因与白细胞基因组甲基化的分子特征(在线方法)密切相关( 第页 2 = 0.59, 对 < 2.2×10 -16 , 图2e ),但仅与组织学检查的污染估计值弱相关( 第页 2 =0.1, 对 = 2.4×10 -12 ; 在线方法; 图2e x轴刻度, 补充图4 ).
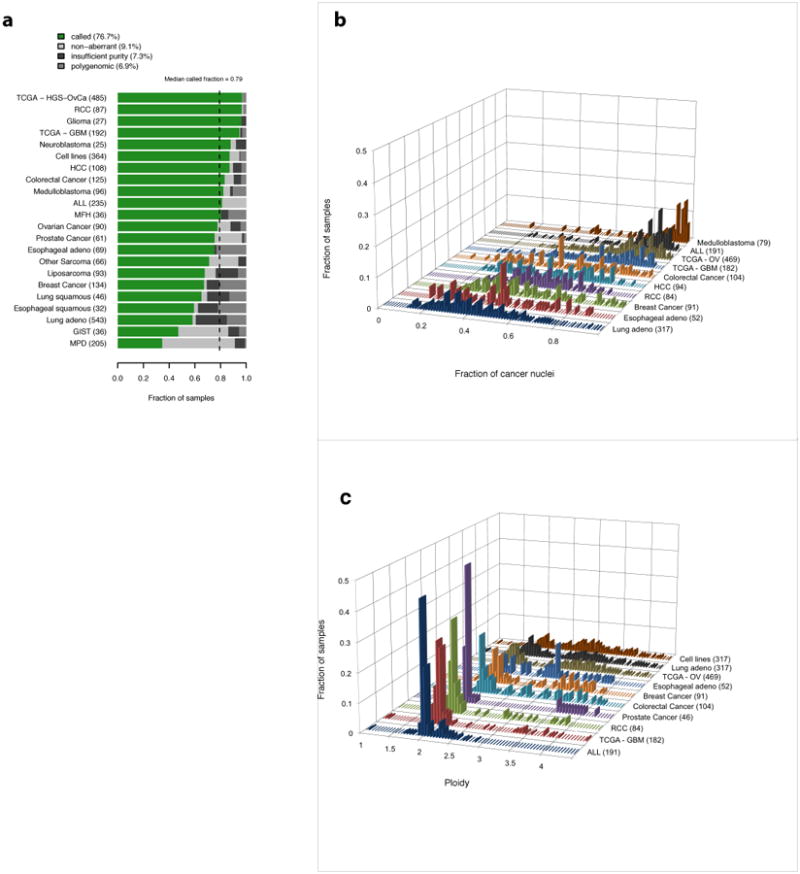
不同癌症类型肿瘤纯度和倍性的评估 我们使用ABSOLUTE分析了来自3155份癌症样本(包括2791份组织样本和364份癌细胞株)的SNP阵列的等位基因拷贝比谱。 这些样本来自两项描述胶质母细胞瘤的TCGA先导性研究(GBM;192个样本) 21 和卵巢癌(488个样本) 33 以及从之前的泛广告文案分析中合并的2445个简介 35 (在线方法)。 少数样本(519或16.4%)无法进行分析,因为它们缺乏可明确识别的SCNA,或者是因为它们接近整倍体(“非异常”),或者是被正常细胞过度污染(“纯度不足”)( 图3a ). 尽管体细胞点突变的测序数据可能解决了这些病例,但该队列中的大多数样本都没有这些数据 35 .
图3。 ABSOLUTE的泛癌应用。
a、, 绝对结果类型:(i)“称为”——唯一纯度/倍性溶液; (ii)“非异常”——样品没有可检测到的体细胞拷贝数变化; (iii)“纯度不足”——癌细胞比例不足; (iv)无法确定“多基因”离散复制比率水平。 参见联机方法和 补充图5 获取每个结果类型的描述和示例。
b、, 几个数据集的估计肿瘤纯度分布。 括号中显示了每组肿瘤样本的数量。 我们注意到,由于使用ABSOLUTE很难调用严重污染的肿瘤,因此其中一些分布偏向于高纯度样本。
c(c) ,括号中显示了每组肿瘤样本的数量。 因为没有SCNA的肿瘤不能用ABSOLUTE来命名,所以这些分布不包括此类样本的流行率。
对于2636个具有可检测SCNA的样本,ABSOLUTE为92%的病例提供了纯度和倍性要求,并将其余样本指定为“多基因”(基因组异质性)( 图3a ),(在线方法; 补充图5 ). 呼叫样本的比例因疾病类型而异,从34.6%(骨髓增生性疾病;主要是非异常基因组)到96.7%(卵巢癌,100%异常基因组),平均呼叫率为79.2%( 图3a ).
估计纯度的分布因癌症类型而异,测试的肺癌、食管癌和乳腺癌样本的平均纯度最低( 图3b ). 污染的影响在不纯肿瘤类型的拷贝率中很明显( 补充图6 ). 估计倍性的分布( 图3c )与之前获得的每种肿瘤类型的细胞学数据定性一致 13 .
每个肿瘤样本的特征以及预测纯度/倍性值的表格如下 补充表1 。另一个表格详细列出了每个肿瘤的分段绝对等位基因拷贝数,如下所示 补充表2 .
通过测序检测体细胞点突变的能力 肿瘤纯度和倍性都会影响检测点突变所需的局部测序深度。 例如,假设在一个含有50%正常细胞污染的样本中,一个区域存在于6个拷贝中,只有1个拷贝携带突变。 在这种情况下,该位点的8个等位基因中只有1个(6个来自癌细胞,2个来自正常细胞)携带突变( 补充图7a ). 因此,我们预计只有12.5%的读操作会观察到突变。 假设测序错误率为10,考虑到这一等位基因部分,需要33倍的局部序列覆盖率才能以80%的灵敏度检测突变 -3 每基和假阳性率控制在<5×10 -7 ,(在线方法, 公式9 , 补充图7b ).
利用ABSOLUTE对纯度和全基因组整数拷贝数的估计,我们可以计算出对每个癌细胞特定等位基因多重性的突变进行有力检测所需的覆盖率。 类似的考虑也适用于通过使用分数多重性检测存在于一部分癌细胞中的亚克隆突变( 补充图7c ). 我们注意到,在设计测序实验的功率计算时,优先考虑以细胞单位表示的肿瘤纯度,而不是DNA分数,因为许多感兴趣的体细胞改变预计会在单个拷贝中发生 每个癌细胞 .
我们分析了等位基因拷贝数分析的癌症样本中纯度和倍性值的分布 35 , 21 , 33 为了确定检测克隆突变所需的适当测序覆盖深度,每个样本的幂为0.8。 为此,我们计算了在给定样本纯度的情况下,在平均拷贝数下,在一个位点检测一个拷贝中的突变所需的读取次数。 (人们可以在拷贝数分布上选择一个特定的百分位。)对于这样的位点,我们发现30倍的局部覆盖率足以满足大多数样本( 补充图7d ). 相比之下,在子克隆中以20%的频率携带突变的平均拷贝数的位点需要覆盖约100倍才能在大约一半的样本中检测到( 补充图7e ). 利用这些计算和基因组局部覆盖率的分布(取决于特定的测序技术),人们可以确定在预定的基因组部分获得足够功率所需的平均覆盖率(例如,在>80%的基因组中>80%的功率)。
然后我们检查了214例TCGA卵巢癌样本的全基因组测序数据(~150×平均覆盖率) 33 确定检测能力是否与实际观察到的突变数量相关。 对于每个样本,我们计算了局部覆盖率提供至少80%的检测能力的位点比例,以检测5%存在的亚克隆中单个拷贝的突变。 这些基因座比例最低的样本往往是2个检测到最少突变的样本( 第页 2 = 0.24, 对 = 2.7×10 -13 ; 补充图7f )这表明,未能找到这种突变是由于缺乏能量。 这一结果也证明了功率计算对亚克隆频谱表征的重要性。
体细胞点突变的多重性分析 接下来,我们使用ABSOLUTE将突变的等位基因片段转换为细胞多样性。 为此,我们检测了29268个在Illumina全杂交捕获测序中发现的体细胞突变 36 214对卵巢癌与正常卵巢癌的数据 33 (在线方法, 图4a ). 肿瘤纯度、倍性和绝对拷贝数值是从Affymetrix SNP6.0杂交数据中获得的,该数据与测序的同一DNA小份相同,允许将等位片段重新标度为多重单位( 图4a ,b; 在线方法, 等式12 ).
图4。 通过SNP阵列和全基因组测序数据的综合分析表征卵巢癌的亚克隆进化。
a、, 214份初级HGS-OvCa样本中检测到29628个体细胞点突变的等位基因分数(交替/总读取计数)直方图 33 .
b、, 中所示突变的等位基因部分( 一 )转换为每个癌细胞的整数等位基因数的点估计值(细胞多样性; x个 -轴)通过校正样品纯度和局部拷贝数。 使用中定义的模型鉴定亚克隆突变 方程式10 .
c、, 克隆与亚克隆点突变的6个可区分核苷酸替换中的每一个的分数。 灰色实线表示 年 = x个 .RMSE:均方根误差。
d-f, HGS-OvCa样品TCGA-24-1603中不同亚克隆群体的分析(纯度=0.96,倍性=1.75)。
日期:, 具有模型化绝对拷贝数的肿瘤SCNA图谱,如 补充图1c,h 正常同源copy-number=1的区域呈灰色,克隆SCNA呈棕色。 亚克隆SCNA(浅蓝色)出现在几个簇中(箭头)。
e、, 点突变等位基因片段谱。 每条实心曲线对应一个突变,密度根据观察到的等位基因断裂和局部读取深度暗示的后验(Beta)分布(在线方法, 等式12 ). 颜色表示克隆亚克隆的分类程度,如( b条 ). 虚线表示单个后验密度的总和。
(f) ,来自的SCNA( d日 )和点突变( e(电子) )被重新缩放到癌细胞分数的单位。 SCNA和点突变(分别为紫色、蓝色和橙色箭头;参见 d日 ).
该程序确定了卵巢癌样本中普遍存在的亚克隆点突变。 虽然许多突变是围绕整数多重性聚集的,但相当一部分突变发生在每个平均癌细胞的多重性大大低于1拷贝的情况下,这与亚克隆多重性一致( 图4b ).
一些证据支持这些亚克隆突变的有效性,包括Illumina对一个独立的全基因组扩增小份进行重新测序,这证实了它们的存在( 补充图8a、b ),并且它们的等位基因部分对应于亚克隆多重性值( 补充图8c,d ). 此外,克隆和亚克隆突变的突变谱相似(RMSE=0.02, 图4c )符合共同的起源机制。 功率计算表明,这些样本至少有80%的功率用于检测发生在10%至53%癌细胞组分中的亚克隆突变,中位数为19%( 补充图7e ).
在大多数样本中,亚克隆突变的多重性分布相似( 图4b )——当在所有样品(未显示)中汇集时,它在特定于样品的检测极限处迅速增加,然后以近似于0.05至0.5倍多重性范围内指数衰减的方式减少。 相比之下,HGS-OvCa样品TCGA-24-1603( 图4d-f )显示了离散的“宏观亚克隆”的证据。 亚克隆SCNA的重定标( 图4d )和点突变( 图4e )癌细胞分离单位( 图4f )在分数0.2、0.3和0.6附近发现了离散簇( 图4f )这意味着每个簇内的改变可能同时发生在相同的细胞中。 我们注意到,细胞部分的组合总和超过1,这意味着至少有一个检测到的子克隆嵌套在另一个子克隆中。
接下来,我们使用ABSOLUTE分析参考和替代等位基因的多重性,将受影响细胞片段中的点突变分为杂合或纯合突变( 图5a-c ). 我们考虑了最近在这些数据中发现的15个突变基因 33 包括5个已知的肿瘤抑制基因(TSG)和5个癌基因( 图5d ). 已知TSG和癌基因的纯合子突变频率显著不同,TSG纯合子变异比例显著升高( 对 = 0.006, 图5d )癌基因无纯合子突变:( 对 = 0.012, 图5d ). 这一结果提供了证据支持 CDK12型 作为卵巢癌的候选TSG 33 ,自2012年7月起 CDK12型 突变是纯合的( 对 = 6.5×10 -5 ; 图5d ).
图5。 214例原发性HGS-OvCa肿瘤样本体细胞突变的多重性分析分类。
a、, 等位基因浓度比率的经验密度估计,通过等位基因部分乘以该位点的复制比率得到。
b、, 等位基因多重性估计的密度估计,如 图4b ,用于对照突变等位基因。 根据突变和参考等位基因的多重性,将突变分为四类。
c、, 四个突变类别中每一个的等位基因浓度比的密度估计显示在 b条 叠加显示。
日期:, HGS-OvCa中显著复发基因的突变分类谱 33 以及之前在这些数据中观察到突变的几个COSMIC基因。 注意,这里只考虑了个别点突变; 未考虑多重事件导致隐性失活的可能性(复合杂合性)。 1412个至少有5个重复突变基因的基因分类分数直方图。 虚线垂直线表示5 第个 (顶部)和95 第个 每个分布的(其他)百分位数。 未观察到多重性>1的突变 NF1型 (未显示)。
总的来说, TP53型 编码外显子中任何基因的克隆、纯合和多重性>1突变比例最高( 图5e ),明确了HGS-OvCa致癌的关键起始事件 37 直接来自基因组数据并且独立于统计复发分析。
全基因组倍增在人类癌症中频繁发生 对于许多癌症类型,总拷贝数(倍性)的分布是显著的双峰分布( 图3c ),与来源于SKY的染色体计数图谱一致 10 , 13 虽然这些结果与它们的体细胞进化过程中的全基因组加倍一致,但很难排除另一种假说,即高倍性核型的进化是由连续的部分扩增过程引起的 12 .
为了研究基因组加倍,我们使用了 同源的 copy-number信息–即拷贝号, b条 我 和 c(c) 我 每个位点的两个同源染色体片段。 通过查看 b条 我 , c(c) 我 在整个基因组中,我们可以得出关于基因组加倍的推论。 基因组加倍后, b条 我 和 c(c) 我 将是偶数。 区域的单个副本丢失后 b条 我 和 c(c) 我 将保持偶数,但较小的将变为奇数。 事实上,当我们观察高倍样本时,我们发现 b条 我 和 c(c) 我 通常在整个基因组中是均匀的,这与它们是由整个基因组加倍而产生的一致( 补充图9 ). 通过模拟,我们发现由于SCNA在多个独立染色体上以串行方式发生,观察到的轮廓不太可能出现( 对 <1e-3; 在线方法)。
利用这些信息,我们可以将样本分为三组,我们将其解释为对应于癌症克隆进化中的0、1和>1基因组加倍事件。 这三组的模态倍性值分别为1.75、2.75和4.0( 图6a )并通过倍性和平均同源拷贝数不平衡将其分为三个簇( 图6b ). 我们将其解释为SNCA发生净损失的证据,其间散布着基因组加倍。 这一过程导致了加倍克隆的中等倍性值(2.2–3.4N),同源染色体普遍失衡( 图6b ).
图6。 原发性癌症中全基因组加倍事件的发生率和时间。
a、 b、, 倍性估计值来自ABSOLUTE。 平均同源失衡被计算为基因组中每个位置同源拷贝数的平均差异。 根据同源拷贝数推断基因组加倍状态(在线方法, 补充图9 ).
c、, MPD—骨髓增生性疾病,ALL—急性淋巴细胞白血病,GBM—多型胶质母细胞瘤,RCC—肾细胞癌,HCC—肝细胞癌,HGS-OvCa—高级浆液性卵巢癌。
日期:, 杂合性缺失定义为0等位基因拷贝。 扩增被定义为0个基因组加倍样本的>1等位基因拷贝,而1个基因组加倍的样本的>2等位基因副本。 根据每个染色体臂的模式等位基因拷贝数进行呼叫。 虚线表示 年 = x个 .
e、, SCNA被定义为与每个样本的模态绝对拷贝数不同的区域,以自适应分辨率将其分为两部分,以保持每个分格200个SCNA,并通过分格长度重新规范化。 每个箱子中的数值进一步除以每个基因组加倍类别中肿瘤样本的数量,用颜色表示,如 一 。黑线表示斜率=-1。 使用SCNAs 0.5对每个类别独立拟合线性回归模型< x个 <20 Mb。 这导致0、1和>1基因组加倍的拟合斜率值分别为-1.05、-0.96和-0.88(未显示)。
基因组加倍的频率因肿瘤类型而异( 图6c )反映了疾病特异性生物学和临床进展状态的差异。 造血肿瘤(MPD、ALL)几乎没有加倍事件,而GBM、RCC、前列腺癌、各种肉瘤、HCC和髓母细胞瘤都有~25%的加倍发生率。 基因组加倍在上皮性癌中更为常见,结直肠癌、乳腺癌、肺癌、卵巢癌和食管癌的加倍发生率均大于50%( 图6c ). 食管腺癌的加倍发病率最高,这与之前关于巴雷特食管进展不同阶段频繁出现“4N”人群的报道一致 38 , 39 .
特定非整倍体先于基因组加倍 然后我们使用ABSOLUTE推断肿瘤发生中基因组加倍的时间顺序,相对于涉及特定染色体臂的SNCA。 在许多癌症类型中,臂级SCNA的固定发生在基因组加倍之前,因为加倍和非加倍样本的特定臂级SNCA频率相似( 图6d , 补充图10 ).
在GBM样本中,涉及9号和10号染色体的LOH以及7号染色体的扩增发生在同等频率( 图6d ),表明GBM中最常见的广泛SCNA发生在基因组加倍之前。 19号和20号染色体的增益几乎只存在于非加倍样本中,在加倍样本中有几个臂的LOH频率更高( 图6d )这表明这些样本背后还存在其他生物差异。
由于ABSOLUTE无法在没有观察到SCNA的病例中区分二倍体2N和4N,因此我们从分析中丢弃了这些非异常样本( 图3a ). 对于许多肿瘤类型来说,由于加倍后染色体丢失的趋势,这种病例是罕见的( 图3c , 图6a、b , 补充图9 ). 然而,由于确定上的差异,特定癌症亚型的表现可能会有偏差。
与广泛的染色体改变相比,局部SCNA事件在加倍基因组中发生的频率更高( 图6e ). 与以前的报告一致 35 , 40 , 41 ,作为其长度函数的焦点SCNA的观测频率( 我 )遵循幂律缩放: 对 ( 我 )⑪ 我 − α ,用于 我 >0.5兆字节( 图6e ). 基因组加倍与较大的SCNA总数相关,但我们获得了各组α接近1的估计值( 图6e )这表明它们产生的机制并不是很大程度上依赖于倍性。
基因组加倍对卵巢癌进展的影响 接下来,我们试图将卵巢癌的全基因组加倍发生率与其他遗传和临床特征相关联。 基因组加倍样本显示杂合子突变的发生率较高,但校正样本倍性消除了这种影响( 图7a )表明碱基突变率是相等的。 多重性>1的克隆突变在双倍样本中的流行率约为10倍; 其中许多事件可能发生在加倍事件之前。 基因加倍样本的纯合子缺失频率较低( 图7b )克隆纯合子突变率低两倍( 对 =1.55×10 -8 , 图7c ). 我们预计,在基因组加倍之前,在加倍样本中观察到的许多纯合子改变是固定的。
图7。 原代HGS OvCa样本中与基因组加倍的遗传和临床关联。
a-e、, 如图所示,颜色与假定的基因组加倍状态相对应。 重要性代码:**– 对 < 10 -5 , * – 对 <0.05,NS– 对 > 0.05.
a-c、, 指示类别中的突变数量作为基因组加倍的函数。 对 -采用双侧Wilcoxin秩和检验计算值,比较基因组加倍为0和1的样本。 误差条表示平均值的标准误差。
日期:, 对 -数值采用双侧Wilcoxin秩和检验计算。
e、, 对 -使用log-rank检验计算值。
基因组加倍样品中纯合子突变的发生率较低,这可能反映了这样一个事实,即在基因组加倍样品中将突变变为纯合子需要更多的事件(尽管这种影响可能会被部分抵消,但是,由于加倍后遗传不稳定性的可能增加,例如中心体复制 42 ). 这些考虑表明,基因组加倍样本通过不同的轨迹进化,因为在加倍后,肿瘤抑制因子失活的发生频率可能较低。
我们注意到,在15个检测到的点突变中,有13个位于肿瘤抑制因子 NF1型 发生在93个未进行基因组加倍的卵巢样本中( 对 = 0.002; Fisher精确测试),这些突变均为纯合子(未显示)。 这与隐性失活的选择一致 NF1型 是抑癌基因的典型模式。 它还表明,非基因组加倍的卵巢癌样本是通过不同的轨迹进化而来的,而不是加倍样本的前兆。 如果没有,很多 NF1型 在加倍的样本中,突变将是纯合的,多重性>1,如图所示 TP53型 .
最后,我们注意到基因组加倍样本与病理诊断年龄的显著增加相关( 图7d )癌症复发率明显较高( 图7e ).
讨论 我们在这里报道了一种可靠的高通量方法的发展,该方法可以从肿瘤衍生DNA样本中推断绝对同源拷贝数,以及点突变的多重值(absolute)。 可以将绝对延伸到其他类型的基因组改变,例如结构重排和小插入/缺失,尽管这可能需要更长的序列读取以确保准确的序列比对。
对SCNA的绝对分析表明,所分析的许多拷贝数变化在样本中所代表的癌症谱系中是固定的( 图3 ). 通过体细胞点突变(其中许多是以整数倍固定的)在卵巢癌中重现了这一点( 图4b ). 根据点突变的多样性对其进行分类可能有助于区分抑癌基因和基因( 图5d ). 离散肿瘤复制状态、亚克隆结构和基因组加倍状态的知识为进一步重建癌症内部的系统发育关系和特定癌症基因组产生的时间序列奠定了基础 43 , 44 , 45 .
ABSOLUTE为利用基因组测序来检测癌症组织样本中的变异等位基因的研究设计提供了一个框架,该研究基于检测突变的灵敏度的计算,作为样本纯度、局部拷贝数和测序深度的函数( 补充图7 ). 基于SNP微阵列数据的ABSOLUTE产生的肿瘤纯度和倍性估计的高准确性( 图2 )可以确定给定样本所需的测序深度,也可以选择给定固定测序深度的合适样本。 这些考虑对解释亚克隆点突变至关重要( 补充图7f,10 ).
absolute对人类癌症中预测的绝对等位基因拷贝数谱的分析为癌症基因组进化提供了新的线索。 观察到的SCNA剖面( 补充图9 )如前所述,与早期染色体不稳定,随后出现稳定的非整倍体克隆的共同轨迹一致 11 我们的数据进一步表明,基因组加倍发生在已经具有相应肿瘤类型特征的臂级SCNA的癌细胞亚群中。 因此,在加倍和进一步克隆生长之前,这些癌症的基因组是通过染色体臂级分辨率的选择形成的( 图6d , 补充图10 ).
这些发现与早期对原发性乳腺癌FACS/SKY图谱的解释大体一致 46 最近在宏观解剖和倍性分类细胞群的研究中进行了综述 14 和单细胞测序 15 原发性乳腺肿瘤。 我们注意到,这个模型代表了对四倍体是一种 启动 事件 47 , 13 , 26 , 48 , 49 此外,基因组谱系的关联( 图6c )卵巢癌的诊断年龄( 图7d )与最近描述的在培养的小鼠胚胎成纤维细胞中连接端粒危机、DNA损伤反应和基因组加倍的机制一致 48 .
本研究中提出的克隆性分析为癌症临床测序提供了一条前进的道路,并提供了解决最近报道的有关肿瘤内异质性的担忧的方法 50 , 14 , 44 , 15 , 45 , 51 , 52 使用ABSOLUTE进行分析可以确定所有癌细胞中存在的与DNA等分体有关的变化( 图1 )即使这种克隆改变对应于少数观察到的突变。 这些改变是特定癌症的候选致癌因素,可能是首选的治疗靶点。 癌症亚克隆体细胞变化的进一步表征对于理解靶向治疗的可变反应可能非常重要,靶向突变的克隆性可能会影响反应水平。
联机方法 纯度、倍性和绝对体细胞拷贝数的推断 无论是细胞学分析还是细胞学分析,种群都对这种分析提出了挑战 10 和基因组数据 11 支持这一假设,正如从配对原发性和转移性病变中获得的类似SCNA图谱的报告一样,同系物特异性拷贝比(HSCR;两条同源染色体的拷贝比估计值)优先用于ABSOLUTE分析, 和用于本研究中的所有分析。虽然ABSOLUTE可以在总复制率数据(例如来自阵列CGH或低通测序数据)上运行,但我们在此不提供此类结果。 HSCR的使用减少了复制配置文件的模糊性。 例如,不含SCNA的样品的总拷贝率曲线与倍性值1、2、3等相等,但HSCR曲线将排除奇数倍性值,因为这些值与相同的同源拷贝数不一致。 此外,由于亚克隆SCNA通常只影响给定基因组片段中两个HSCR值中的一个,因此当考虑HSCR而非总拷贝数时,克隆与亚克隆SCNAs全基因组的比率通常更高。
HSCR是通过HAPSEG程序对杂合子位点的阶段性多点等位基因复制比进行分段估计得出的 53 Affymetrix SNP阵列数据。 作为该程序的一部分,来自群体连锁分析HAPMAP3的单倍型面板 54 与统计相位软件BEAGLE结合使用 55 为了估计每个癌症样本中SNP标记的阶段性生殖系基因型。 这增加了我们分辨这些基因型的敏感性,因为它自然地利用了SNP之间的局部统计依赖性 53 此外,这允许更大程度地解决同源拷贝率之间的小差异,因为来自SCNA引起的杂合标记的等位基因失衡的相位信息可以与来自单倍型面板的统计相位相结合 53 .
候选肿瘤纯度和倍性值的鉴定和评估 我们描述了候选肿瘤纯度和倍性值的鉴定及其计算 SCNA配合 使用概率模型进行log-likelihood评分。 这是通过用高斯混合模型拟合输入HSCR估计值来实现的,其中成分集中在由 等式1 该模型还支持一小部分不限于离散水平的亚克隆事件。 通过在纯度和倍性值的大范围内搜索此可能性的局部最优值来确定候选解决方案。 这导致了具有相应SCNA-fit可能性的离散候选解集( 等式1 , 补充图1d,h ).
这些分数量化了通过将观察到的HSCR解释为整数SCNA而提供的每个解决方案的证据。 这些计算对于每个样本都是独立的。 输入数据包括 N个 HSCR x个 我 , 我 ∈ {1, …, N个 }. 每一项都有标准误差 σ 我 ,与表示的基因组部分相对应 w个 我 。每个 x个 我 假设是由以下任一原因引起的 问 整数拷贝数状态: 问 = {0,1, …, 问 −1},或附加状态 Z轴 对应于亚克隆拷贝数。 我们将可能的复制状态集合称为 S公司 = 问 ∪ Z轴 。我们定义 问 +1个指示器 秒 对于每个段的复制状态 第页 ( 秒 我 )表示分段概率 我 已从状态生成 秒 ∈ S公司 .的整数copy-states S公司 被编入索引 q个 ∈ 问 ; 非整数状态表示为 z(z) .
每个整数拷贝数对应的预期拷贝比率 q个 ( x个 )肿瘤样本中的 方程式1 注意,当使用同源复制比率时,该等式变为:
由于HSCR是相对于单倍体浓度测量的,而不是由 等式。 1 . D类 与肿瘤纯度和倍性有关( α 和 τ )( 等式1 , 补充图1 ). 观察到的 x个 我 使用以下混合建模 问 高斯分量位于 μ = { μ q个 ∈ 问 }表示整数复制状态 问 和一个额外的统一组件 Z轴 .混合物 Z轴 允许为片段分配非完整复制值,以便偶尔的子克隆更改或伪影不会显著影响可能性。
和 分别表示正常密度和均匀密度。 自由参数 σ H(H) 表示样本级噪声超过HSCR标准误差 σ 我 ,这可能代表了恶性细胞群中的适度数量的相关克隆,持续的基因组不稳定,或由于可变的实验条件而产生的过度噪音。 混合物重量 θ = { θ 秒 ∈ S公司 }指定分配给每个复制状态的预期基因组部分。 参数 d日 表示均匀密度的域,对应于合理的拷贝比值范围(我们使用 d日 = 7 ).
由于数据由基因组分段计算的复制率组成,因此出现了一些复杂情况。 为了一致解释,混合物权重( 对 ( 秒 我 | w个 我 , θ ))必须分别计算每个片段,并考虑可变基因组分数 w个 我 这是通过限制分配给每个复制状态的基因组质量的标准平均值来实现的,以匹配 θ :
其中: 表示所有配置的平均值{ 秒 我 },由函数加权 = 对 ( 秒 我 | w个 我 , λ )该密度对应于最大熵分布 秒 受这些约束:
哪里 秒 # 表示状态的顺序 秒 在复制状态序列中,从0开始。 的值 问 拉格朗日乘数 λ 通过Nelder-Mead优化确定 我 2 损失:
这种近似允许SCNA-fit分数对数据过度分割的鲁棒性。 给定段的可能性 我 然后计算为:
然后,数据的完整对数似然为:
我们定义参数化 b条 = 2(1 − α ), δ τ = α / D类 ,它决定 μ 通过 等式(3) .通过优化 等式(5) 关于 b条 和 δ τ .计算 等式(5) 需要估计 θ 和 σ H(H) ,尚不清楚 先验的 我们做了一个近似(尺度分离),假设 等式(5) 对这些参数的适度波动保持不变。 每种情况的临时可能性 x个 我 然后可以通过以下公式计算
然后通过优化
从跨越域的正则格中的所有点开始 b条 和 δ τ .参数 σ 对 在本研究中设置为0.01。 我们验证了上述近似识别模式与通过全Metropolis-Hastings Markov chain Monte Carlo(MCMC)模拟获得的模式等效(数据未显示)。 近似值允许使用更简单的计算。
每个解决方案的SCNA-fit得分是在优化 σ H(H) :
具有以下元素 θ 计算每个值 σ H(H) 签署人:
每个模式的SCNA-fit对数似然的最终计算通过插入 μ̂ , θ ^ 、和 σ̂ H(H) 进入之内 等式(5) 。每个分段的复制状态指示符的估计计算如下:
请注意,每个 q̂ 我 是一个向量,表示每个变量的后验概率 问 ∈ 问 整数复制状态,对应于复制比率(位置) μ .
相对于DNA倍性估计,基因组范围内的绝对拷贝保护文件被过度确定。 倍性的另一个估计值可以计算为基因组上的预期绝对拷贝数:
根据定义,这个数量( τ̂ 克 )是对癌症倍性的另一种估计(请注意,当使用HSCR时,添加了一个额外的因子2)。 因为( τ̂ 克 )是模型数据中离散状态的加权平均值,预计它对稍微改变或缩放复制文件的实验波动更为稳健。 注意,对于此计算 q̂ ij公司 计算方法为 θ̂ z(z) =0,因此上述期望仅超过整数状态。
我们验证了这些估计值通常接近( τ̂ )通过优化SCNA-fit似然(RMSE=0.26, 补充图12a ). 然而,我们注意到倍性估计值和校准数据平均值之间的不一致程度之间的关系( 补充图12b ). 注意到正确校准的复制比率数据的平均值始终为1,我们检查了校准错误是否是由于数据中的缩放偏差造成的。 我们发现,该模型解释了两个估计之间近三分之二的不一致性(校正的RMSE=0.09, 补充图12c )由此我们推断,标度偏差主导了我们的校准错误。 这很重要,因为这些偏差不会影响肿瘤纯度的估计( 补充图12 ).
复制状态位置的两个附加转换 μ 当使用微阵列测量复制率时使用。 其中第一个解释了等温吸附模型的衰减效应 7 :
其中,值 ϕ̂ 参数化给定样本中的衰减响应,并通过HAPSEG进行估计。 第二种转换是根据 56 :
哪里 σ η 和 σ ε 表示每个微阵列的乘性和加性噪声等级,由HAPSEG估计。 在估计 x个 我 值,之后它们的分布近似正态。 中规定的正常混合物成分( 4 )然后变成 小时 ( x个 我 ) = 小时 ( 克 ( μ q个 ))+ ε 我 ,并在这些变换下执行相应的似然计算。
核型模型 为了从通过拟合模型确定的候选组中可靠地选择正确的肿瘤纯度和倍性解决方案,通常需要附加信息( 4 ). 在给定的肿瘤样本中,理论上可能的纯度、倍性和拷贝数值的几种组合可能映射到等效拷贝比( 补充图1c,h ). 此外,亚克隆SCNA的存在可能会导致虚假的高倍性解决方案,通过过度离散拷贝配置文件,使难以置信的核型获得更大的SCNA-fit可能性,从而允许将其分配到整数拷贝级别( 附图1h-j ).
ABSOLUTE根据肿瘤组绝对同源拷贝数分布的相似性对肿瘤组进行分组,从而对常见癌症核型进行建模( 补充图2 ). 这些模型是以“boot-strapping”的方式直接从肿瘤数据构建的,其中使用具有相对明确轮廓(例如,由于高纯度值)的肿瘤子集初始化模型,迭代地允许调用更多肿瘤等。 以前人类癌症的细胞遗传学特征被用来指导这一过程 13 。这些模型可以计算 核型可能性 ,对于每个候选纯度/倍性溶液,反映了相应核型与输入肿瘤样本的特定疾病相关模型的相似性( 8 ). 结合SCNA-fit和核型可能性有助于在许多肿瘤样本中准确鉴定纯度和倍性值( 补充图1d,h ). 选择一种不太常见的核型的溶液需要来自SCNA拷贝图谱的更多证据。
特定疾病的核型特征的先验知识总结为以下内容的混合 K(K) 整数同调复态上的多元多项式分布 问 =每个染色体臂[0,7]。 对于给定的候选纯度和倍性解决方案,每个片段对应的片段复制状态指标 我 , qâ ij公司 ,总结为 J型 臂级同源拷贝数,表示 Ĉ 核型对数似然分数计算如下:
哪里 w个 我 表示每种混合物成分的重量。 核型模型 K(K) 我 是 J型 × 问 使用标准期望最大化(EM)算法对模型副本文件的臂级同源副本状态进行聚类,得到SCNA概率矩阵 57 对于多项式混合物。 该计算确定了具有相似基因组拷贝谱的疾病亚型组( 补充图2 ). 注意,每个臂的两个同源物的复制状态都是建模的( J型 = 78). 使用两条同源染色体的多项式概率卷积计算只有总复制率数据的样本的核型得分。
集群数量 K(K) 通过最小化贝叶斯信息准则(BIC)复杂性惩罚来选择每种疾病:-2 L̂ k个 + 科威特 日志( N个 ),其中 L̂ k个 表示ℒ的总和 K(K) 值超过 N个 输入样本,使用计算 K(K) 集群。 为了避免局部极小,EM算法对每个值运行25次 K(K) ∈[2,8],起始点随机,保留最佳模型。
这些模型是以半自动化的方式构建的,通过植入相对明确的防拷贝文件。 随着肿瘤的增加,重复核型的使用清楚地确定了额外样本的正确解决方案等。例如,chr17的LOH发生在几乎100%的卵巢癌样本33中,这使得模型能够了解到,暗示chr17 LOH的解决方案可能是正确的。 总共创建了14种疾病类型的模型。 ABSOLUTE称之为样本少于40个的疾病在此过程中被忽略。 此外,通过合并所谓的原发性癌症特征,创建了一个“主”模型。 该模型用于没有特定核型模型的疾病。
从复制文件数据进行联合纯度/倍性推断的局限性 将SCNA-fit和核型模型精确校准到数据所暗示的真实确定性水平,将允许为每个候选解决方案分配概率; 我们认为,我们在这里提出的模型没有充分捕捉到癌症基因组的复杂性,无法进行这样的解释。 即使是人工审查,使用ABSOLUTE进行分析有时也可能导致错误的解释,例如,在没有随后可检测到的增益或损失的情况下,基因组加倍可能会导致一个包含真实倍性值一半的解决方案,这在某些情况下可能对应于合理的核型模型。 或者,当多个亚克隆SCNA出现在相邻克隆峰之间的中点附近时,可以选择一个意味着真倍性加倍的解决方案。 我们注意到,在我们的框架中无法调用没有可靠检测到SCNA的样本(倍性2N或4N;纯度待定)。 因此,这些样品被排除在下游分析之外(见下文)。 推断错误率的估计需要独立测量样本倍性。 对不同肿瘤类型的进一步验证实验将有助于澄清任何特定疾病的警告。
我们注意到,体细胞突变等位基因部分的使用,结合SCNA拷贝率,通常可以提高SCNA较少的样本的灵敏度。 此外,突变数据有助于区分纯度/倍性估计中的基因组加倍模糊性,尽管它们没有告知类型的模糊性 b′ = b条 + 2(1-α)/ D类 ( 补充图1d,i , 等式1 ). 因此,组合分析通常有助于使用ABSOLUTE(未显示)获得更高的呼叫率。
幸运的是,我们的泛癌SNP阵列数据集中的许多样本在基因组加倍之前和之后都与频繁的SCNA保持一致,从而能够在不使用体细胞点突变数据的情况下对许多样本进行明确的推断。 癌症基因组进化的这一方面先前在乳腺癌细胞遗传学数据46中有所记录。 我们注意到,在生成FACS验证数据或分析NCI60细胞系倍性估计值之前,对ABSOLUTE结果进行了手动审查( 图2a、b ).
鉴定不符合纯度/倍性推断的样品 为了便于对本研究中使用的许多癌症样本进行快速分析,ABSOLUTE被编程为自动识别无法可靠调用的拷贝特征,并将其分类为信息性故障类别( 图3a ),由以下标准定义。定义 米 作为后向全基因组复制状态分配的排序向量( θ̂ ),因此 米̂ 1 代表了 θ̂ (模态复制状态)。 该向量由 θ 0 替换为0,如果 θ 0 <0.01和 b条 <0.15,因此种系拷贝数变体(CNV)或遗传纯合子区域不会与小SCNA混淆,这意味着样本非常纯。 然后分类如下:
非异常: 米̂ 三 < 0.001, 米̂ 2 < 0.005, σ̂ H(H) < 0.02
纯度不足: 米̂ 三 < 0.001, 米̂ 2 <0.005时, σ̂ H(H) ≥ 0.02
多基因的: θ̂ z(z) > 0.2.
这些标准适用于每个样本的顶级模式(结合SCNA-fit和核型得分)。 每个结果的几个示例如所示 补充图5 上述指定使得自动呼叫与手动审查后获得的呼叫具有相当好的一致性。 我们注意到,体细胞点突变数据的使用增加了这些样本类别中的呼叫敏感性。
癌细胞系DNA混合实验 从两个癌细胞株(HCC38、HCC1143)提取的DNA与匹配的B淋巴细胞株(HCC38BL、HCC1144BL)以不同比例混合,并与Affymetrix 250 K Sty SNP阵列杂交。 通过将DNA浓度标准化至50ng,为每个细胞系创建DNA储备小份/ μ l.按体积将癌症和匹配的B淋巴细胞DNA混合到每个所需的混合分数。
原发性肿瘤样本的FACS分析 卵巢浆液性癌患者的福尔马林固定和石蜡包埋块可从肿瘤切片中获得,对应于从中获得DNA等分样品用于SNP阵列杂交的冷冻块。 将含有至少70%肿瘤细胞核的多个卷曲切割成150微米的总厚度。 切片被分解并用碘化丙啶标记(DNA染色)。 FACS用于测定倍性。
通过病理学检查确定肿瘤纯度 从多家医院的组织库中收集冷冻卵巢浆液性囊腺癌标本,并将其保存在液氮蒸汽中。 用两侧的H&E载玻片(任意命名为顶部和底部)制作组织部分,如下所示:将组织安装在最佳切割温度介质(OCT)中,并将其加热至-20°C。 答4 μ m冰冻切片(顶部幻灯片)用低温恒温器切割(Leica Microsystems,Wetzlar,德国)。 通过用手术刀从组织表面刮下100毫克肿瘤组织,然后再刮下4毫克,制作出一个用于分子提取的样本 μ m冻结段被切割(底部滑动)。 使用Autostainer XL和一体式拖鞋(徕卡)对两个玻片组织切片进行H&E染色。 使用Scanscope XT(Aperio,Vista,CA,USA)以20倍分辨率创建幻灯片的数字图像。 由董事会认证的病理学家通过ImageScope软件(Aperio)远程进行病理学审查。 病理学家最初在低倍镜下检查每张幻灯片,以确定低倍镜的形态,然后将放大倍数增加到20倍,并在每张幻灯片上检查10个具有代表性的高倍镜场。 卵巢浆液性囊腺癌的诊断得到了证实,肿瘤纯度是指肿瘤细胞核与载玻片上总细胞核的比例。 提取标本的肿瘤纯度计算为顶载玻片和底载玻片的平均纯度分数。 质量控制包括由第二位病理学家随机检查10%的载玻片,以验证读数的一致性。
白细胞甲基化特征 489例高分期、高级别浆液性卵巢肿瘤和8例正常输卵管样本的DNA甲基化数据来自 http://tcga.cancer.gov/dataportal/ 此外,还获得了两名女性的浅黄色皮毛样本。 所有数据均使用Illumina Infinium HumanMethylation 27阵列生成,该阵列查询了位于NCBI数据库(Genome Build 36)中14475个一致性编码序列转录起始位点附近的27578个CpG位点。 每个探针的DNA甲基化水平总结为β值范围从0(未甲基化)到1(甲基化) 58 .
白细胞甲基化特征推导如下。 每个探针都根据浅黄色皮毛和输卵管样本中平均β值的差异进行排序。 我们保留了100个在正常输卵管组织和外周血白细胞中平均DNA甲基化之间具有最大正差异和最大负差异的探针,指定 不列颠哥伦比亚省 和 英尺 (分别富含浅黄色皮毛和输卵管)。 让 T型 伊克 表示探针的β值 k个 在肿瘤样本中 我 .让 B类 k个 表示每个探针的棕黄色涂层样品的平均β值。 让 T型 k个 表示所有肿瘤样本中观察到的最小β值 不列颠哥伦比亚省 探针和最大值 英尺 探针。 表示方式 (f) B类 样品中棕黄色涂层成分的分数,则每个探针的方程如下: T型 伊克 = B类 k个 (f) B类 + T型 k个 (l− (f) B类 ). 求解此等式 (f) B类 给出: (f) B类 = ( T型 伊克 − T型 k个 )/( B类 k个 − T型 k个 ). 的值 (f) B类 对签名中的200个探针中的每一个进行了计算,并获得了核密度估计值。 然后计算白细胞特征作为这种密度估计的模式。
数据集的选择 我们分析了2445个Affymetrix 250 K Sty SNP样本,这些样本来自之前的泛癌调查 35 包含3131个癌症样本。 因为我们的数据处理需要使用鸟食算法 59 ,无法使用缺乏二倍体PCR对照的外部数据集。 此外,样本少于20个的癌症类型被排除在外。 此外,从TCGA GBM中采集了680个Affymetrix SNP6.0样本 21 和HGS-OvCa 33 研究,以及30个细胞系样本,使总样本数达到3155。 完整的癌症样本分析表如下 补充文件1 。ABSOLUTE结果的完整表格如下所示 补充文件2 .
癌症组织样本体细胞突变检测的功率计算 我们开发了一个用于检测突变的统计能力计算框架。 检测变异的能力取决于等位基因分数 (f) 和局部覆盖深度 n个 为了计算功率,我们对随机序列错误与速率一致的理想场景进行建模 ε 。我们计算支持读取的最小数量 k个 这样 k个 或由于排序错误导致的更多相同的非参考读取小于定义的假阳性率(FPR):
在哪里?
≥的变量 k个 然后认为检测到支持读取。 我们指定了测序错误率 ε = 1 × 10 −3 且FPR=5×10 −7 用于本研究中的所有计算。 功率计算如下:
在哪里?
我们考虑在癌组织衍生DNA样本中检测每个癌细胞单个拷贝处存在的克隆体细胞变体的情况。 给出纯度估计值( α )和本地绝对拷贝数( q个 吨 ),这些变体的等位基因部分为:
功率是在如下情况下计算的( n个 , δ ).
为了简化功率和肿瘤纯度/倍性之间的关系 补充图7 ,我们考虑了预期基因座在全基因组拷贝平均值上的检测能力。 功率由样品决定 等位指数δ τ = α / D类 ,这仅仅是肿瘤纯度/倍性的函数( 等式。 1 ). 通过使用等位基因分数获得期望功率 (f) = δ τ 在里面 等式(9) 此计算仅在替换预期基因组拷贝数方面有所不同,即倍性( τ )对于本地copy-number q个 吨 在里面 等式:(10) .
分数形式的预期亚克隆变异体的功率 秒 (f) 的癌细胞是由Pow提供的( n个 , 秒 (f) δ τ )此计算用于 补充图7c,e 使用Pow进行本地副本计算( n个 , 秒 (f) δ )用于 补充图7f、11 .
卵巢癌体细胞点突变的检测 我们分析了全基因组杂交捕获Illumina测序(WES) 36 TCGA联合会先前分析的214对卵巢癌肿瘤-正常对照的数据 33 我们使用了程序muTect(K.Cibulskis等人,正在准备中)。我们使用了比以前分析该数据时使用的程序muTett更新的版本 33 新版本的主要改进是减少了先前体细胞突变在0.5的等位基因部分的情况,允许在低等位基因部分的突变(例如不纯样品中的克隆事件)或对亚克隆突变更敏感。 该过程导致29268个体细胞突变。
点突变多重性的推断 基于肿瘤纯度和全基因组绝对拷贝数的知识,我们开发了一个概率模型,用于推断种系和体细胞变体的整数多重性。 将突变位点的绝对同源拷贝数表示为 q个 1 和 q个 2 ,带有 q个 1 ≥ q个 2 。种系变体的可能多样性如下:
哪里 q个 吨 = q个 1 + q个 2 假设所有的体细胞点突变都是在单个单倍型上唯一出现的,那么可能的多重性为:
请注意,当只有总复制比率数据可用时, q个 2 上述情况未知,并且 q个 吨 而是使用。
种系突变通常存在于癌细胞和正常细胞群体中,体细胞拷贝数的改变会影响等位基因部分。 种系中的杂合变体,具有多重性 克 q个 在癌症基因组中,具有等位基因部分:
其中,纯合生殖系变体的等位基因分数为1,与 α 对于体细胞点突变,多重性下的预期等位基因分数 秒 q个 是 (f) 秒 q个 = 秒 q个 δ ,带有 δ 如中所示 方程(10) .
考虑一个观察到的未知拷贝的体细胞点突变 秒 q个 ∈ 秒 q个 ,观察到的等位基因分数 fè 、和 n个 覆盖轨迹的总读数。 完全可能 f̂ 可以表示为对应于每个元素的β分布的混合 秒 q个 ,再加上一个附加组件 S公司 对应于亚克隆状态:
哪里 w个 S公司 q个 ∈ w个 q个 指定每个状态的混合物重量 秒 q个 、和 w个 S公司 c(c) 指定子克隆组件权重。 亚克隆成分 S公司 由贝塔分布(建模采样噪声)与亚克隆癌细胞分数的指数分布组成,具有单个参数 λ :
注意指数分量中坐标的变化,使用 δ ; 这使得无论肿瘤纯度和局部拷贝数如何,都可以用一致的癌细胞分数单位进行建模(注意,这种分布在单位间隔上是重新规范化的)。 给定整数复制状态的概率 秒 q个 然后可以计算为:
类似地,给定突变为亚克隆的概率计算如下:
对于本研究中的计算,我们修正了 λ = 25, w个 秒 q个 至0.25,以及 w个 S公司 c(c) 到0.75,这与组合样本突变分数分布相吻合( 图4b ). 结果显示于 图4 对各种设置都很稳健。
与整数体细胞多重性对应的混合组分权重的优化可以通过与SNCA混合模型中描述的方法类似的方式完成 等式(6) Dirichlet先验可以指定为伪计数向量,其值等于每个重数值的先验观测值。 然后根据观测计数计算出的后Dirichlet模式计算重量。 当使用成对SCNA和体细胞点突变数据运行ABSOLUTE时,这些计算用于计算每个纯度倍性模式的可能突变分数。
支持基因组加倍推断的癌基因进化模拟 进行了简单的模拟,以获得 对 -观察到的同源拷贝数配置可能由独立增益和损耗的串行过程产生的概率值。 基因组范围的同源拷贝数在染色体臂分辨率下总结为整数增益/损耗(共78个状态)。 然后我们确定收益/损失的总数 N个 对于样本,并计算每个手臂的速率,这些速率被归一化为概率。 通过独立采样进行样本模拟 N个 从这些概率中获得和损失。 每个样本重复1000次,记录次数 米 在观察到的样品中,甚至达到或超过了较高的同源拷贝数。 这个 对 -值为: 对 = 米 /1000,如果 米 >0,否则 对 < 0.001.