总结
肝脏的化学或创伤性损伤通常与异常愈合(纤维化)有关,后者会抑制肝脏再生1–5在肝脏修复过程中,肝脏小生境细胞差异调节再生和纤维化的机制尚待确定6–8肝血管生态位主要由肝窦内皮细胞(LSEC)代表,部署旁分泌滋养因子,称为血管分泌因子,以刺激再生9–15然而,目前尚不清楚LSEC的促再生血管分泌信号是如何被破坏以促进纤维化的16,17这里,通过结合诱导性内皮细胞(EC)特异性小鼠基因缺失策略和急性和慢性肝损伤的互补模型,我们揭示了来自LSEC的不同血管分泌信号在即刻损伤后诱导再生,并在慢性损伤后引发纤维化。血管生态位的原纤维化转变是由基质衍生因子-1(SDF-1)受体、CXCR7和CXCR4的差异表达引起的18–21在LSEC中。急性损伤后,LSEC中CXCR7上调与CXCR4共同作用,诱导转录因子Id1,部署促再生血管分泌因子并触发再生。可诱导删除抄送7成年小鼠LSEC(抄送7iΔEC/iΔEC)通过减少Id1介导的血管分泌因子的产生而损害肝脏再生9–11相反,在反复注射肝毒素(四氯化碳)和胆管结扎造成慢性损伤后,LSEC中的组成性FGFR1信号抵消了CXCR7依赖的促再生反应和CXCR4表达的增加。CXCR4在CXCR7表达上的优势改变了LSEC的血管分泌反应,刺激结蛋白的增殖+肝星状细胞22,23加强原纤维血管生态位。EC-特定消融Fgfr1级(Fgfr1级iΔEC/iΔEC)或Cxcr4号机组(Cxcr4号机组iΔEC/iΔEC)在小鼠中恢复了促再生途径,并阻止了FGFR1介导的血管分泌因子的适应不良颠覆。同样,LSEC中选择性CXCR7激活可消除纤维生成。因此,我们已经证明,在应对肝损伤时,血管生态位中促再生CXCR7/Id1和促纤维化FGFR1/CXCR4血管分泌途径的差异招募平衡了再生和纤维化。这些结果为实现肝再生而不引起纤维化提供了治疗路线图1,2,4.
尽管肝脏具有再生能力,但慢性或过度损伤往往会导致肝纤维化,最终导致肝硬化和肝衰竭1–7肝脏修复的综合过程包括以细胞外基质(ECM)蛋白的合成为特征的再生和伤口愈合。这两个过程都受到实质性肝细胞和非实质性细胞(NPC)之间动态相互作用的调节7,22,24,25包括肝星状细胞1,23、炎症细胞6,8胆道上皮细胞和肝窦内皮细胞9,13,15,16因此,定义平衡再生和功能失调(适应不良)愈合的多细胞串扰5有望为肝脏疾病设计治疗方案。
排列肝窦血管的LSEC以超出其代谢产物传递被动作用的方式诱导肝器官发生9,12,14,15通过部署旁分泌生长调节剂(我们将其定义为血管分泌因子),LSEC触发肝细胞再生9–11,25,26然而,在慢性损伤的情况下,LSEC的异常激活会导致纤维化16,17LSEC在介导肝脏修复中的生态位功能的这种二分法表明,不同的血管分泌信号平衡再生和纤维化11因此,我们试图破译将LSEC的促再生能力破坏为促再生状态的机制。
作为对组织损伤的反应,细胞因子和趋化因子,如基质衍生因子(SDF)-1(Cxcl12)上调,通过其受体CXCR4和CXCR7启动再生18–21虽然造血细胞和血管细胞中的CXCR4激活都调节血管生成和造血,但另一种SDF-1受体CXCR7的表达主要局限于内皮细胞,其功能主要被认为在血管模式和肿瘤新生血管生成中起关键作用。然而,在明确的肝损伤环境中缺乏细胞类型特异性遗传模型,阻碍了SDF-1途径协调肝脏修复的机制的阐明。
为了揭示LSEC在调节肝脏修复中的不同作用,我们采用了一次性注射四氯化碳(CCl4)以及导致急性肝损伤的对乙酰氨基酚,以及反复CCl的慢性损伤模型4注射和胆管结扎(图1a). 值得注意的是,在单次CCl后的第2天4损伤,CXCR7在VE-cadherin中特异性上调+LSEC公司(图1b-e,补充图1). 相反,CXCR4在其他细胞类型中广泛表达,CCl后其在LSEC上的表达相对稳定4受伤。因此,我们已经确定CXCR7是一种诱导性LSEC特异性SDF-1受体,用于肝脏损伤反应。
图1。急性肝损伤后,肝窦内皮细胞(LSEC)中基质衍生因子(SDF)-1受体CXCR7的上调可诱导血管分泌介导的再生。
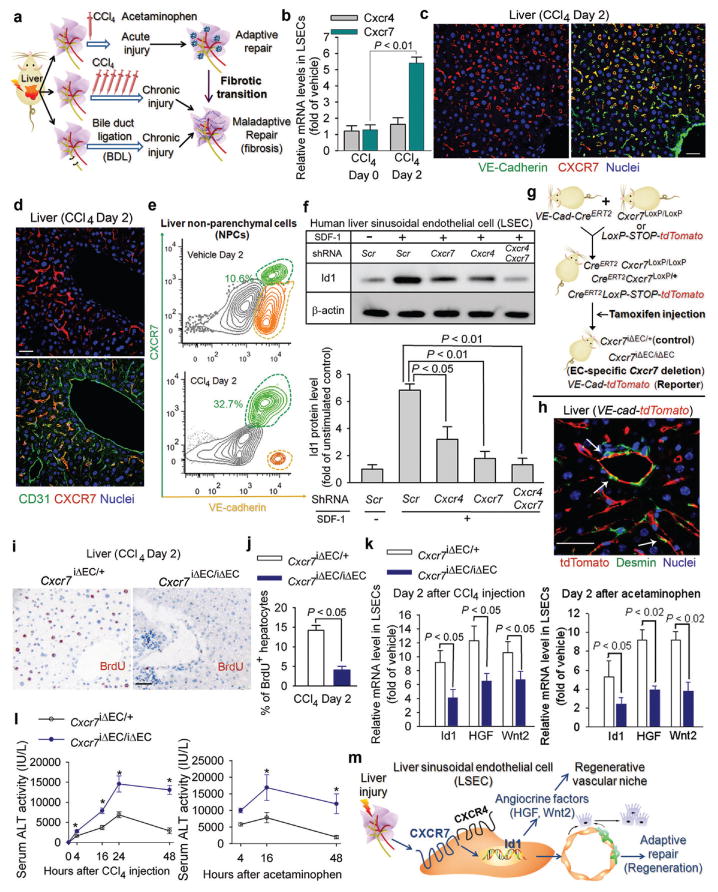
a) 肝损伤模型,用于研究促再生LSEC功能向促纤维化血管生态位的不适应过渡。
b至e)CXCR7在VE-cadherin上特别上调+CD31型+急性化学损伤后的LSEC。注入四氯化碳(CCl)后4),在分离的LSEC(b)、肝切片(c,d)和非实质细胞(NPC)(e)中测定CXCR7和CXCR4。CXCR7在LSEC上表达,但在大血管上不表达;N=5。图1中的比例尺=50μm,以下所有数据均表示为平均值±标准平均误差(s.e.m.)。
f) 刺激人LSEC的SDF-1上调DNA结合抑制剂1(Id1),这是一种诱导产生促再生血管分泌因子的转录因子9人原代因子VIII中SDF-1对Id1的刺激+LSEC因沉默而被废除Cxcr4号机组和抄送7在LSEC中;N=5。
g、 h)内皮细胞(EC)特异性诱导缺失抄送7(抄送7iΔEC/iΔEC)在小鼠中。窝藏老鼠液氧磷站点-侧面抄送7与具有EC特异性的小鼠系杂交VE-卡德林启动子驱动的CreERT2段(VE卡芯T2段). 的特异性VE卡芯T2段在携带td番茄蛋白的报告小鼠中验证了该方法。抄送7三苯氧胺注射液诱导内皮细胞缺失或td番茄表达9.抄送7iΔEC/+小鼠作为对照。注意内皮细胞中tdTomato的特异表达,而不是结蛋白+星形细胞(h,白色箭头)。
i-l)肝再生受损和肝损伤加重抄送7iΔEC/iΔEC小鼠急性肝损伤后。通过BrdU掺入染色(i,j)测定细胞增殖。CCl后LSECswas中Id1和促再生血管分泌因子、肝细胞生长因子(HGF)和Wnt2的表达4用扑热息痛(k)和血清丙氨酸氨基转移酶(ALT)水平测定肝损伤程度(l);N=5。
m) LSEC中CXCR7的激活触发Id1介导的促再生血管分泌因子的产生。急性肝损伤后,CXCR7与CXCR4协同作用,在LSEC中诱导前再生Id1通路,并触发血管分泌介导的肝再生。
我们的团队9,10和其他13已有研究表明,肝部分切除后,LSEC通过产生血管分泌因子,如Wnt2和肝细胞生长因子(HGF),诱导肝再生。LSEC中转录因子Id1的激活对这一过程至关重要9SDF-1在培养的人类LSEC中诱导Id1上调,这一上调被两种基因沉默所消除抄送7或Cxcr4号机组(图1f,补充图2、3). 值得注意的是,CXCR7-选择性激动剂TC14102同样诱导Id1上调。免疫沉淀Western blot(IP-WB)表明,在SDF-1刺激后,CXCR7与LSEC中的CXCR4和β-抑制素相关(补充图4). 因此,SDF-1通过CXCR7和CXCR4之间的合作刺激Id1诱导27,28.
为了确定CXCR7在LSEC介导的肝脏修复中的作用,我们使用他莫昔芬诱导的EC特异性CreERT2系统系统拆除抄送7在成年小鼠的内皮细胞中(图1g). 窝藏老鼠LoxP公司场地两侧抄送7与VE卡芯ERT2系统EC-特异性小鼠VE-卡德林促进剂驱动Cre公司ERT2系统使用携带td番茄荧光蛋白的小鼠,用漂浮的终止密码子来排除VE卡芯ERT2系统其他肝细胞类型。特异性激活的三苯氧胺注射液Cre公司ERT2系统内皮细胞中的活性,但不表达去糖蛋白的星状细胞(图1h,补充图5),证明诱导的EC特异性缺失抄送7(抄送7iΔEC/iΔEC)英寸VE卡-Cre公司ERT2系统抄送7液氧磷/液氧磷老鼠。抄送7-单倍体成年小鼠(抄送7iΔEC/+)用作Cre毒性的对照。
与对照小鼠相比抄送7iΔEC/iΔECCCl后小鼠数量显著减少4受伤(图1i,j). 血管分泌因子HGF和Wnt2的Id1依赖性部署抄送7iΔEC/iΔEC两次CCl后,小鼠数量减少4和对乙酰氨基酚引起的肝损伤(图1k). 血清丙氨酸氨基转移酶(ALT)水平测定的肝损伤程度加重(图1l,补充图6). 因此,肝损伤后,SDF-1通过CXCR7的激活+LSEC触发血管分泌反应以启动肝脏再生(图1m).
虽然肝细胞在急性肝损伤后再生,但慢性肝损伤更频繁地导致肌成纤维细胞的明显激活并导致纤维化2,三,5为了解决LSEC的促再生血管分泌信号如何被转移以刺激这种不适应的愈合,我们采用了重复CCl的慢性小鼠肝损伤模型4注射29(图2a、b). 值得注意的是,慢性损伤后CXCR4上调可抵消LSEC中的CXCR7-Id1通路(图2c-e,补充图7、8). 重复CCl后4注射后,α-平滑肌肌动蛋白(SMA)和ECM蛋白胶原的蛋白水平在抄送7iΔEC/iΔEC小鼠与对照小鼠的比较(图2f-h,补充图9-10). 值得注意的是,注射CXCR7-特异性激动剂TC14102降低了对照组SMA和I型胶原的上调,但没有抄送7iΔEC/iΔEC老鼠(图2g-h,补充图11). 因此,慢性肝损伤会干扰LSEC中的促再生CXCR7-Id1血管分泌途径,并促进纤维化。
图2。反复肝毒性损伤扰乱了LSEC中CXCR7促再生途径,并迫使产生促纤维化血管生态位。
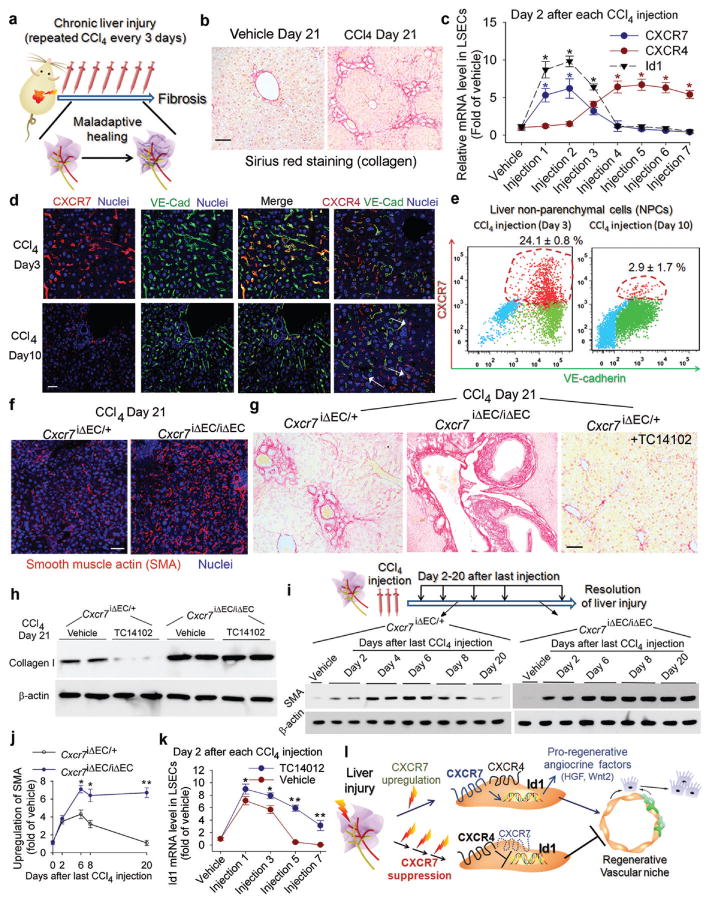
a-b)反复注射CCl诱导小鼠肝纤维化429天狼星红染色检测损伤肝脏中的胶原蛋白。图2中的标度br=50μm。
c-e)慢性肝损伤抑制CXCR7通路并上调LSEC中CXCR4的表达。定量PCR(c)、免疫染色(d)和流式细胞术(e)显示VE-cadherin(VE-Cad)中CXCR7-Id1通路消失+慢性CCl后的LSEC4受伤。CXCR4以EC和非EC表示(白色箭头)。*,P(P)<0.05,与车用小鼠相比;N=8。
f-h)LSEC中CXCR7激活可消除肝纤维化。纤维化程度在抄送7iΔEC/iΔEC小鼠肝脏α-平滑肌肌动蛋白(SMA)和I型胶原水平升高。值得注意的是,CXCR7-选择性激动剂TC14012减少了对照组的纤维化,但没有抄送7iΔEC/iΔEC老鼠;N=7。
i、 j)肝脏损伤的解决受损抄送7iΔEC/iΔEC老鼠。测试受损肝脏中的SMA水平,以评估损伤的解决情况(i)。与对照组小鼠相比,SMA水平抄送7iΔEC/iΔEC小鼠在最后一次CCl后增强4注射后保持稳定。胶原蛋白I水平也进行了类似评估(补充图13); *,P(P)< 0.05, **,P(P)<0.01,与对照组小鼠相比;N=5。
k) CXCR7激活可恢复慢性损伤LSEC的Id1诱导。TC14102在重复CCl期间阻止LSEC中Id1抑制4受伤。*,P(P)< 0.05, **,P(P)与车辆组相比,<0.01;N=7。
l) LSEC中促再生CXCR7-Id1通路的干扰导致血管生态位的促纤维化转变。损伤后,LSEC中CXCR7-Id1通路的上调诱导肝活性血管分泌因子的生成并刺激再生。慢性损伤扰乱CXCR7-Id1信号传导,阻碍再生并刺激纤维化。
检测LSEC中CXCR7在解决肝纤维化方面的需求6,23.三次CCl后4注射后,对照组小鼠的SMA和胶原蛋白水平增强,并在最后一次注射后的第8天达到峰值,在第20天接近基础水平(车辆注射组)(图2i-j,补充图12-13). 相比之下,肝损伤的时间依赖性分辨率在抄送7iΔEC/iΔEC老鼠。重复注射CCl抑制Id1通路4由CXCR7激动剂TC14102诱导(图2k). 因此,在对损伤的反应中,LSEC中的CXCR7通路在刺激再生和解决纤维化方面发挥着不可或缺的作用。反复刺激后,CXCR4通路优于CXCR7-Id1通路导致纤维化(图2l).
然后我们采用胆管结扎术(BDL),一种临床相关的肝胆汁淤积模型,来确定CXCR4和CXCR7如何调节LSEC的原纤维化转变(图3a). BDL会导致胆道上皮损伤,并导致持续胆汁淤积和肝硬化。BDL后,LSEC通过窦周结蛋白进行投资+星状细胞(图3b). 类似于重复的CCl4损伤后,LSEC中CXCR4暂时上调,CXCR7-Id1通路受到抑制(图3c). 值得注意的是,SMA沉积在抄送7iΔEC/iΔEC小鼠高于对照组抄送7iΔEC/+老鼠(图3d-f). 因此,在BDL诱导的胆汁淤积性损伤期间,LSEC中CXCR7信号的丢失导致纤维化。
图3。LSEC中FGFR1过度激活导致的胆汁淤积性肝损伤将CXCR7依赖的促再生反应转移到CXCR4主导的促纤维化血管生态位。
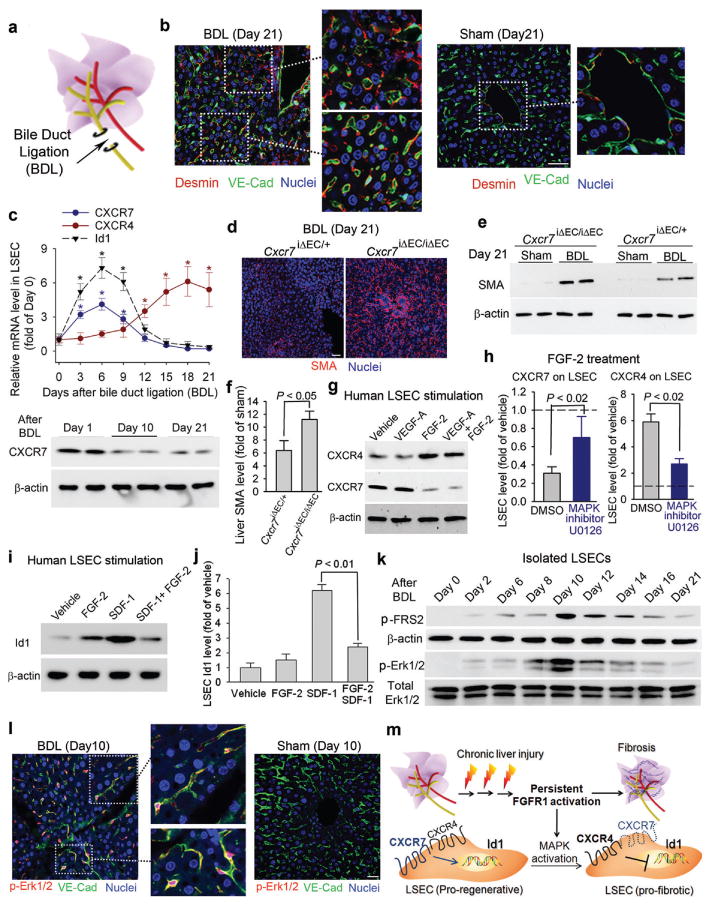
a、 b)胆管结扎(BDL)诱导的胆汁淤积性损伤导致LSEC的前纤维化转变。BDL诱导的胆汁淤积性肝损伤(a)后,大多数VE-cadherin+LSEC被窦周结蛋白覆盖+成纤维细胞(b,插图)。相比之下,desmin+假手术肝中星状细胞稀疏分布。比例尺=图3中的50μm。
c) BDL抑制CXCR7-Id1通路并上调LSEC中CXCR4的表达。底部面板显示了BDL后LSEC中CXCR7蛋白的丢失;*,P(P)与第0天的水平相比,<0.05;N=5。
d-f)BDL引起的肝纤维化在抄送7iΔEC/iΔEC老鼠。BDL之后,抄送7iΔEC/iΔEC小鼠肝脏SMA蛋白水平高于对照组;N=4。
g-j)FGF-2通过MAP激酶激活促进LSEC中CXCR4信号传导,抵消CXCR7-Id1通路。FGF-2(而非VEGF-A)上调CXCR4,抑制CXCR7,并抑制LSEC中SDF-1依赖性Id1的诱导。这种FGF-2介导的CXCR4相对于CXCR7的优势被MAP激酶(MAPK)抑制剂U0126减弱;N=5。
k、 l)BDL后LSEC中FGFR1和MAPK通路的激活。VE-cadherin中FGFR1下游效应器FRS2(p-FRS2)和Erk1/2(p-Erk1/2)的磷酸化/活化呈时间依赖性增强+BDL后的LSEC;N=6。
m) LSEC中通过MAPK激活的组成性FGFR1信号迫使CXCR4主导的原纤维化血管生态位。在慢性肝损伤期间,FGFR1介导的LSEC异常MAPK激活上调CXCR4并干扰CXCR7-Id1通路。LSEC中FGFR1/CXCR4激活的优势决定了从适应性(促再生)到不适应性(促纤维化)肝脏修复的病理进展。
然后用血管生成因子VEGF-A和FGF-2刺激人LSEC,以研究CXCR4表达增强以主导CXCR7途径的机制。FGF-2而非VEGF-A诱导CXCR4 mRNA和蛋白水平,并减弱CXCR7表达(图3g,补充图14). 值得注意的是,MAP激酶的特异性抑制(MAPK阻断了LSEC中FGF-2驱动的CXCR4诱导和CXCR7抑制11(图3h). 因此,FGF-2联合SDF-1治疗人类LSEC可阻断SDF-1单独诱导Id1(图3i-j,补充图15)表明FGF-2在LSEC中诱导CXCR4上调和CXCR7抑制,从而抵消Id1促再生途径。
为了测试FGF-2信号如何调节LSEC的血管分泌反应,我们检测了BDL后LSEC上FGF-2受体FGFR1的激活。FGFR1呈时间依赖性上调和激活16,9,30损伤VE-cadherin中MAP激酶(Erk1/2)磷酸化+LSEC公司(图3k,l,补充图16-17). 因此,胆汁淤积性损伤导致LSEC中FGFR1介导的MAPK激活,导致肝脏修复期间CXCR4主导的血管分泌反应的促纤维化转变(图3m).
为了阐明LSEC生态位原纤维化漂移的机制,我们有条件地消融Fgfr1级和Cxcr4号机组成年小鼠的内皮细胞(Fgfr1级iΔEC/iΔEC和Cxcr4号机组iΔEC/iΔEC) (图4a). BDL后,结蛋白的正弦周膨胀+细胞、胶原和SMA沉积、MAPK激活和CXCR7抑制Fgfr1级iΔEC/iΔEC与对照组小鼠相比,所有小鼠均减毒(图4b-g,补充图18). 值得注意的是,CXCR7依赖的Wnt2和HGF的血管分泌表达在Fgfr1级iΔEC/iΔEC老鼠(图4e). 因此,EC特异性删除Fgfr1级在成年小鼠中,BDL阻止了LSEC向促纤维化状态的异常转变。
图4。LSEC中CXCR4的FGFR1激活在肝脏修复中激发促纤维化血管分泌信号。
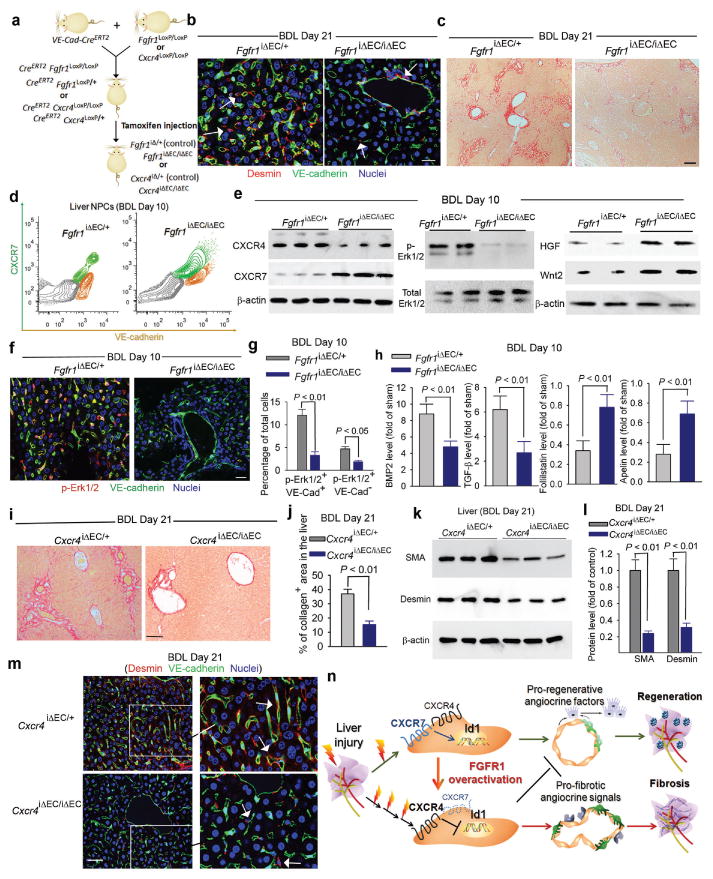
a) EC特异性诱导缺失Fgfr1级和Cxcr4号机组(Fgfr1级iΔEC/iΔEC和Cxcr4号机组
iΔEC/iΔEC)成年小鼠。
b、 c)减少肝纤维化Fgfr1级iΔEC/iΔEC老鼠。与对照小鼠相比,窦周结蛋白富集+肝内星形细胞(b,白色箭头)和胶原沉积(c)减少Fgfr1级iΔEC/iΔECBDL后小鼠;N=4;比例尺=图4中的50μm。
d-g)EC特定删除Fgfr1级在小鼠中,可防止CXCR4介导的BDL后的适应不良转变,并恢复再生血管分泌信号。在BDL损伤的LSEC中Fgfr1级iΔEC/iEC小鼠,VE钙粘蛋白中CXCR7的抑制和CXCR4的上调(d)和Erk1/2的激活+LSEC(e-g)减少。伴随着肝活性血管分泌因子HGF和Wnt2的恢复生成;N=4。
h) LSEC中敏捷因子的原纤维生成减少Fgfr1级iΔEC/iΔEC老鼠。BDL刺激LSEC中血管分泌因子的不同生成,包括BMP和TGF-β途径中因子的上调,以及抗纤维化基因如卵泡抑素和apelin的抑制(补充图19). BDL后LSEC中血管分泌因子的促纤维化漂移被缓解Fgfr1级iΔEC/iΔEC老鼠;N=4。
i-m)减少肝纤维化Cxcr4号机组iΔEC/iΔECBDL后的小鼠。BDL后肝纤维化程度在Cxcr4号机组iΔEC/iΔEC与对照组小鼠相比,小鼠的胶原蛋白(i,j)、SMA(k,l)沉积减少,窦周结蛋白富集+星状细胞(m,白色箭头);N=5。
n) 来自LSEC的发散性血管分泌信号平衡肝脏再生和纤维化。急性肝损伤后,LSEC中CXCR7-Id1通路的激活刺激肝活性血管分泌因子的产生。相比之下,慢性损伤会导致LSEC中FGFR1的明显激活,从而干扰CXCR7-Id1通路,并有利于CXCR4驱动的促纤维化血管生成素反应,从而引发肝纤维化。因此,为了应对损伤,差异启动的LSEC部署不同的血管分泌信号以平衡肝脏再生和纤维化。
为了揭示慢性损伤的LSEC中血管分泌反应的改变,我们从BDL和假手术小鼠中分离并分析了LSEC(补充图19). 在受损的LSEC中,促纤维化因子(包括TGF-β、BMP2和PDGF-C)显著上调,同时抗纤维化基因(如卵泡抑素和apelin)受到抑制。BDL后LSEC中血管分泌因子产生的这种差异性漂移在Fgfr1级iΔEC/iΔEC小鼠,如抗纤维化基因的恢复和纤维化因子表达的减少(图4h).
BDL后的纤维化程度在Cxcr4号机组iΔEC/iΔEC老鼠。与对照组相比Cxcr4号机组iΔEC/+小鼠、SMA和胶原的肝脏沉积以及结蛋白的窦周堆积+星形细胞减少Cxcr4号机组iΔEC/iΔECBDL后的小鼠(图4i-m,补充图20). 减少肝纤维化Cxcr4号机组iΔEC/iΔEC小鼠暗示,慢性损伤导致的LSEC中组成性CXCR4激活建立了原纤维血管龛,激活了邻近的肌成纤维细胞并引发纤维生成(图4n).
虽然肝部分切除术后的肝脏再生进行得无可挑剔,没有纤维化,但慢性损伤后的肝脏修复与纤维化有关,纤维化会损害肝功能的恢复。因此,识别调控肝再生和异常愈合的分子途径将为肝硬化和肝衰竭的治疗开辟新的途径。我们已经证明,70%的肝部分切除术后,LSEC中VEGFR2-Id1通路的激活可导致肝再生9在这里,我们证明了FGFR1介导的LSEC中CXCR4上调和CXCR7抑制抵消了LSEC的促再生功能并导致纤维化。
我们采用互补的急性和慢性损伤模型来解读LSEC在肝脏修复中的作用(图1a). 在小鼠肝脏中,我们已经确定了对急性损伤作出反应的LSEC中促再生CXCR7依赖性信号的优先诱导(图1). 在慢性肝损伤期间,LSEC中CXCR7的缺失和CXCR4的上调会导致纤维化的进展。事实上,CXCR7激活在促进再生和对抗纤维化方面的关键功能可以通过选择性激活LSEC中的CXCR7减轻纤维化以及受损的再生来证明(图1)和增强纤维生成(图2–三)英寸抄送7iΔEC/iΔEC老鼠。
LSEC中SDF-1信号从CXCR7依赖的促再生反应向CXCR4主导的促纤维化功能的转变是由于持续的FGFR1驱动慢性MAPK激活11,16,29,30(图3). 我们利用一个可诱导的EC特异性小鼠基因缺失系统来证明FGFR1-CXCR4通路在LSEC的原发性漂移中的作用。值得注意的是,慢性肝损伤后,LSEC中CXCR4相对于CXCR7的表达增强在Fgfr1级iΔEC/iΔEC老鼠,以及两者Fgfr1级iΔEC/iΔEC和Cxcr4号机组iΔEC/iΔEC小鼠对纤维化有抵抗力(图4). 因此,损伤后LSEC中CXCR7-Id1的激活触发并保护肝源性血管分泌因子的产生,而慢性刺激引起的显性FGFR1-CXCR4激活将LSEC转化为促纤维化生态位。通过FGFR1-CXCR4途径,LSEC的异常血管分泌功能导致结蛋白的激活和扩张+星状细胞。阐明肝星状细胞如何相互调节LSEC在肝脏修复中的表型和功能贡献仍有待研究1,23此外,LSEC上CXCR7和CXCR4的激活是否调节炎症反应,例如巨噬细胞的募集,这些巨噬细胞可能调节肝再生和纤维化6,8,需要研究。
总之,我们发现差异激活的LSEC为肝脏修复提供不同的血管分泌信号。LSEC中CXCR7的选择性激活有助于引导血管分泌介导的再生。通过组成性FGFR1激活对CXCR7通路的干扰将LSEC中的SDF-1信号转移到不适应(促纤维化)的血管分泌反应。识别在肝血管生态位中协调不同血管分泌反应的分子途径将为确保肝脏修复而不导致纤维化的治疗策略奠定基础。
方法
内皮细胞(EC)特异性基因缺失策略
通过治疗可诱导EC特异性基因缺失VE-Cadherin-CreER公司T2段用三苯氧胺包庇小鼠31.Cxcr4号机组LoxP/LoxP之前描述过老鼠32. TheCre公司+小鼠接受三苯氧胺250mg/kg腹腔注射,连续6天,第三次给药后中断3天。三苯氧胺治疗三周后,通过定量PCR和免疫印迹分析证实LSEC中的靶基因缺失。所有动物实验都是根据动物保护和使用委员会制定的指导方针进行的,实验对象是性别/年龄/体重相匹配的同窝动物。
肝损伤和纤维化模型
单次和多次注射CCl4如前所述,分别用于诱导急性和慢性肝损伤29.CCl公司4在油中稀释,最终浓度为40%(0.64mg/ml),并以1.6 mg/g体重注射到小鼠体内。对8至10周龄小鼠进行胆管结扎(BDL)。腹腔注射400 mg/kg对乙酰氨基酚也可诱导小鼠急性肝损伤。为了进行BDL,小鼠在全身麻醉下接受一个3厘米长的腹部中部切口。在距离肝门约1cm的两个相邻位置结扎总胆管。然后在结扎的两个部位之间切开导管。
为了选择性激活CXCR7,CCl后将激动剂TC14102(R&D Systems,MN)腹腔注射到小鼠体内4每隔一天以30 mg/kg的剂量对损伤或BDL损伤进行一次。在指定的时间点,处死小鼠并采集整个肝组织进行纤维生成分析,包括天狼星红染色的I型胶原沉积和免疫印迹检测的SMC和I型胶原蛋白沉积(Abcam,CA)。
小鼠肝细胞的分离培养
如前所述,通过改良的两步胶原酶灌注技术从小鼠中分离肝细胞9简单地说,用肝脏灌注培养基(Invitrogen,CA)灌注肝脏,并用肝脏消化培养基(Initrogene,CA)分离。非实质细胞(NPC)用75%percoll储备液和35%percoll储存液进行percoll梯度离心分离。通过小鼠LSEC结合磁珠(Miltenyi,CA)和Dynabeads®磁珠结合抗鼠VEGFR3抗体(Imclone,NY)分离LSEC9从分离的LSEC中测定Id1、CXCR4、CXCR7和FGFR1蛋白和mRNA的表达11为了检测LSEC中的FRS-2 phsophorylation,在收获组织之前用磷酸酶抑制剂灌注小鼠(Pierce,CA)11.
人类LSEC的培养和刺激
人类LSEC从Science Cell Inc.获得,并按照供应商的指示进行培养。通过免疫染色或流式细胞术分析验证CXCR7、VE-粘附素、vWF和因子VIII的表达。选择性击倒Cxcr4、Cxcr7在LSEC中,通过共转染15μg穿梭慢病毒载体(含有目标基因或用钙沉淀法在293T细胞中打乱shRNA、3μg pENV/VSV-g、5μg pRRE和2.5μg pRSV-REV。病毒上清液通过超速离心浓缩并用于转导人类LSEC。
为了测定细胞因子刺激后LSEC中Id1、CXCR4和CXCR7的表达,500000个LSEC被播种并用Cxcr4号机组,抄送7或搅乱shRNA慢病毒。在无血清培养基中饥饿后,用10 ng/ml SDF-1或20 ng/ml FGF-2刺激种子LSEC。在不同的时间点,收集细胞以测量Id1蛋白和mRNA的表达。用30μM U0126处理以抑制MAPK的活性。使用抗p-Erk1/2和总Erk1/2抗体(细胞信号技术,MA)的免疫印迹分析MAPK(p-Erk1/2)的激活11.
对于免疫沉淀(IP)-Western blot(WB),细胞裂解物通过带有蛋白酶抑制剂鸡尾酒和磷酸酶(Pierce)的RIPA裂解缓冲液回收,并与与蛋白A/G珠(Invitrogen,CA)结合的抗CXCR7抗体(R&D Systems,MN)孵育。在归一化为细胞裂解液中的CXCR7总蛋白量(输入)后,用磁铁取回小球,洗脱相关蛋白,并通过Western blot(加州圣克鲁斯)测定β-arrestin、CXCR4和CXCR7的相关性。
肝NPC和LSEC的流式细胞术分析
为了进行流式细胞术分析,将从处死动物中取出的肝脏切碎,在肝脏消化介质(Invitrogen,CA)中消化,并通过30μm的过滤器过滤。用Fc阻断剂(CD16/CD32;BD Biosciences,CA)预先阻断单细胞悬液,然后与以下识别小鼠LSEC和造血细胞的主要抗体孵育:大鼠IgG2aκ和IgG2a-β同型对照;CD31/PECAM-1(克隆MEC 13.3,eBioscience,CA);VE-cadherin/CD144(克隆Bv13,eBioscience,CA);CXCR7(克隆11G8,明尼苏达州研发系统公司)。通常,根据制造商的说明,使用抗体标记试剂盒(Invitrogen,CA)将初级抗体直接与各种Alexa荧光染料或量子点结合。在Alexa Fluor 750的情况下,使用琥珀酰酯进行偶联,并通过BioSpin P30凝胶(Bio-Rad)进行纯化。
用LSRII流式细胞仪(Beckton Dickenson)测量标记细胞群;用FACS-Diva软件对多变量实验进行补偿。使用多种对照,如同种型抗体和未染色样品进行流式细胞术分析,以确定多变量流式细胞术中所需的适当门、电压和补偿。
肝脏冷冻切片的免疫染色和组织学分析
为了获取组织进行组织学分析,用4%PFA灌注小鼠,冷冻保存,并在OCT中快速冷冻。对于免疫荧光(IF)显微镜,阻断肝脏切片(10μm)(5%驴血清/0.3%Triton X-100),并在原始抗体中孵育:抗VE-卡德林多克隆抗体(pAb,2μg/ml,R&D Systems,MN),抗CD31单抗(MEC13.3,5μg/ml,BD Biosciences,CA)、抗CXCR7单抗(11G8,5μg/ml,R&D Systems,MN)、抗结蛋白(pAb,2μg/ml;Abcam,CA)和抗p-Erk1/2抗体(2μg/ml,Cell Signaling Technology,MA)。在荧光团结合二级抗体(2.5μg/ml,Jackson ImmunoResearch,PA)中孵育后,用TOPRO3或DAPI(Invitrogen,CA)对切片进行复染。
肝细胞增殖体内通过BrdU摄取量测量。简言之,小鼠在死亡前60分钟(50 mg/kg)腹腔注射单剂量BrdU(Sigma)。取下肝叶,称重并进一步处理。切片在室温下用1 M HCl预培养1 h,在室温下使用10 mM Tris(pH 8.5)中和15 min,并使用BrdU检测系统(BD Biosciences,CA)和荧光结合二级抗体(2.5μg/ml,Jackson ImmunoResearch,PA)进行染色。为了免疫组织化学(IHC)检测BrdU,在肝脏冷冻切片上进行内源性过氧化物酶和非特异性蛋白块(5%BSA、10%驴血清和0.02%吐温-20),并与次级pAb和链霉亲和素辣根过氧化物酶孵育(Jackson ImmunoResearch,PA)。
图像采集和分析
用Olympus BX51显微镜(Olympus America,NY)捕获肝载玻片的IHC染色,并在AxioVert LSM710共焦显微镜(Zeiss,NY)上记录荧光图像。此外,还测定了VE-cadherin与CXCR4和CXCR7的共训练。
实时聚合酶链反应分析基因表达
使用RNeasy试剂盒从冷冻保存的肝组织或分离的LSEC中提取总RNA(加州齐根市)。分离后,使用上标逆转录酶试剂盒(Invitrogen,CA)将500 ng总RNA转录成cDNA。使用定量聚合酶链反应(Applied Biosystems)检测特定基因的cDNA表达。
致谢
我们感谢Timothy Hla博士批判性地阅读了这份手稿,感谢Rob Berahovich博士和Kevin Eggan博士对CXCR7信号转导用于肝脏修复的建议。获得美国心脏协会国家科学家发展基金(#12SDG1213004)和纽约干细胞基金会Druckenmiller Fellowship的支持。S.R.由Ansary干细胞研究所支持;霍华德·休斯医学研究所;帝国州立干细胞委员会和纽约州卫生部拨款(NYSTEM,C024180,C026438,C026878);NHLBI R01s HL097797、DK095039和HL119872;卡塔尔国家重点研究基金会NPRP08-663-3-140和卡塔尔基金会生物医学研究计划(BMRP)。
脚注
作者贡献:B.-S.D.和Z.C.构思了这个项目,进行了实验,并撰写了论文。R.L.、D.N.和P.G.进行了实验并分析了数据。M.E.P、M.S.、K.S.和S.Y.R.解释了数据。S.R.设计了项目,分析了数据并撰写了论文。所有作者都对手稿发表了评论。
工具书类
-
1Friedman SL、Sheppard D、Duffield JS、Violette S。纤维化疾病的治疗:接近起跑线。科学转化医学。2013;5:161sr167。doi:10.1126/scitranslmed.30004700。[内政部] [公共医学] [谷歌学者]
-
2Bataller R,Brenner DA。肝纤维化。临床投资杂志。2005;115:209–218. doi:10.1172/JCI24282。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
三。Iredale JP。肝纤维化模型:探索炎症和实体器官修复的动态本质。临床投资杂志。2007;117:539–548. doi:10.1172/JCI30542。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
4Gurtner GC、Werner S、Barrandon Y、Longaker MT。伤口修复和再生。自然。2008;453:314–321. doi:10.1038/nature07039。[内政部] [公共医学] [谷歌学者]
-
5韦恩助教。在各种纤维增生性疾病中,共同和独特的机制调节纤维化。临床投资杂志。2007;117:524–529. doi:10.1172/JCI31487。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
6Duffield JS等。巨噬细胞的选择性耗竭揭示了肝脏损伤和修复过程中不同的相反作用。临床投资杂志。2005;115:56–65. doi:10.1172/JCI22675。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
7迪尔AM。邻里守望组织协调肝脏再生。《国家医学》2012;18:497–499. doi:10.1038/nm.2719。[内政部] [公共医学] [谷歌学者]
-
8Boulter L等。巨噬细胞衍生Wnt反对Notch信号传导,以确定慢性肝病中肝祖细胞的命运。《国家医学》2012;18:572–579. doi:10.1038/nm.2667。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
9Ding BS等。肝再生需要来自窦内皮的诱导性血管分泌信号。自然。2010;468:310–315. doi:10.1038/nature09493。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
10丁伯生,等。内皮源性血管分泌信号诱导和维持再生肺泡极化。单元格。2011;147:539–553. doi:10.1016/j.cell.2011.10.003。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
11Kobayashi H等。来自Akt激活内皮细胞的血管紧张素因子平衡造血干细胞的自我更新和分化。自然细胞生物学。2010;12:1046–1056. doi:10.1038/ncb2108。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
12Sakaguchi TF、Sadler KC、Crosnier C、Stainier DY。内皮信号调节斑马鱼肝细胞顶叶极化。当前生物量。2008;18:1565–1571. doi:10.1016/j.cub.2008.08.065。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
13王磊,等。肝窦内皮细胞祖细胞促进大鼠肝再生。临床投资杂志。2012;122:1567–1573. doi:10.1172/JCI58789。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
14Matsumoto K、Yoshitomi H、Rossant J、Zaret KS。内皮细胞在血管功能之前促进肝脏器官发生。科学。2001;294:559–563. doi:10.1126/science.1063889。[内政部] [公共医学] [谷歌学者]
-
15LeCouter J等人,《血管生成诱导的肝依赖性内皮保护:VEGFR-1的作用》。科学。2003;299:890–893. doi:10.1126/science.1079562。[内政部] [公共医学] [谷歌学者]
-
16Huebert RC等。水通道蛋白-1促进肝硬化病理性新生血管系统的血管生成性侵袭。肝病学。2010;52:238–248. doi:10.1002/hep.23628。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
17Zeisberg EM等。内皮细胞向间充质细胞的转化导致心脏纤维化。2007年《国家医学》;13:952–961. doi:10.1038/nm1613。[内政部] [公共医学] [谷歌学者]
-
18Miao Z等。CXCR7(RDC1)促进体内乳腺和肺肿瘤生长,并在肿瘤相关血管中表达。美国国家科学院院刊2007;104:15735–15740. doi:10.1073/pnas.0610444104。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
19Yu S、Crawford D、Tsuchihashi T、Behrens TW、Srivastava D。趋化因子受体CXCR7起调节心脏瓣膜重塑的作用。开发动态。2011;240:384–393. doi:10.1002/dvdy.22549。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
20Sierro F等。第二个CXCL12/SDF-1受体CXCR7缺陷小鼠的心脏发育受到干扰,但造血功能正常。美国国家科学院院刊2007;104:14759–14764. doi:10.1073/pnas.0702229104。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
21Tachibana K等。趋化因子受体CXCR4对胃肠道血管化至关重要。自然。1998;393:591–594. doi:10.1038/31261。[内政部] [公共医学] [谷歌学者]
-
22Armulik A、Genove G、Betsholtz C.周细胞:发育、生理和病理角度、问题和承诺。开发单元。2011;21:193–215. doi:10.1016/j.devcel.2011.07.001。[内政部] [公共医学] [谷歌学者]
-
23Troeger JS等。小鼠肝纤维化消退过程中肝星状细胞的失活。胃肠病学。2012;143:1073–1083. doi:10.1053/j.gastro.2012.06.036。e1022。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
24Zaret KS,Grompe M.肝脏和胰腺细胞的生成和再生。科学。2008;322:1490–1494. doi:10.1126/science.1161431。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
25.Woo DH等。人类胚胎干细胞衍生的肝细胞样细胞对小鼠肝脏修复的直接和间接贡献。胃肠病学。2012;142:602–611. doi:10.1053/j.gastro.2011.11.030。[内政部] [公共医学] [谷歌学者]
-
26Hoehme S等人。预测和验证微血管上的细胞排列作为恢复肝脏再生中组织结构的顺序原则。美国国家科学院院刊2010;107:10371–10376. doi:10.1073/pnas.0909374107。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
27Decaillot FM等。CXCR7/CXCR4异二聚体组成招募β-抑制素以增强细胞迁移。生物化学杂志。2011;286:32188–32197. doi:10.1074/jbc。M111.277038。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
28Rajagopal S等。Beta-arrestin-而非G蛋白-通过“诱饵”受体CXCR7介导的信号传导。美国国家科学院院刊2010;107:628–632. doi:10.1073/pnas.0912852107。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
29.Yu C,等。成纤维细胞生长因子1型和2型在四氯化碳诱导的肝损伤和纤维化形成中的作用。《美国病理学杂志》。2003;163:1653–1662. doi:10.1016/S0002-9440(10)63522-5。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
30Bohm F等。FGF受体1和2控制小鼠再生肝脏的化学诱导损伤和复合解毒。胃肠病学。2010;139:1385–1396. doi:10.1053/j.gastro.2010.06.069。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
31Wang Y等。Ephrin-B2控制VEGF诱导的血管生成和淋巴管生成。自然。2010;465:483–486. doi:10.1038/nature09002。[内政部] [公共医学] [谷歌学者]
-
32Nie Y等。CXCR4在维持外周B细胞亚群和体液免疫中的作用。实验医学杂志。2004;200:1145–1156. doi:10.1084/jem.20041185。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。

