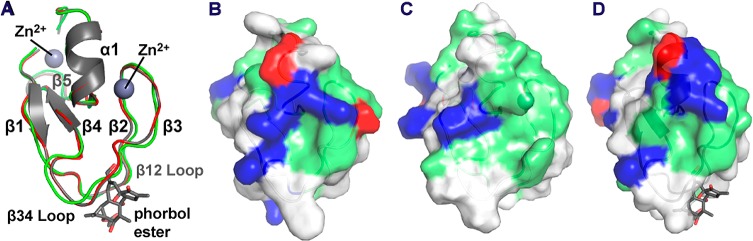
介绍 当从 细胞溶质到膜,许多信号 蛋白质被激活以响应脂质活化剂与 它们的膜结合域。 1 − 三 作为膜结合的范例 结构域,C1结构域在许多方面发挥着重要的调节作用 信号蛋白如蛋白激酶C(PKC)、蛋白激酶 D(PKD)、二酰甘油激酶(DAGK)和嵌合蛋白。 4 − 8 每个C1域包含约5条短交错链和一条 C-末端螺旋,围绕两个完整的Zn组织 2+ 离子(图 1 A) ●●●●。 按C1的顺序 域,一个特征基序,HX 10–12 CX公司 2 CX公司 9–15 CX公司 2 CX公司 4 高x 2–4 CX公司 6–8 C、 已确定,其中H和C代表 保守的组氨酸和半胱氨酸残基与锌配合 2+ 离子,X可以是任何氨基酸。 4 , 9 , 10 据信,疏水性的一半 C1结构域,主要由β12和β34组成 环,穿透膜的碳氢化合物核心, 2 , 11 , 12 而亲水性的一半 锌 2+ 表面的离子和几个离子残留物 暴露于胞浆中(图 1 B–D)。
图1。
(A) 叠合结构 PKCαC1A结构域的同源性 模型; 红色)、PKCαC1B域(PDB代码:2ELI;绿色)和 配体结合的PKCδC1B结构域(PDB编码:1PTR;灰色)。 C类 α PKCαC1A模型之间的RMSD为0.1Å 和PKCδC1B结构,以及PKCα 和PKCδC1B结构。 (B)的曲面表示 PKCαC1A结构域,(C)PKCαC1结构域,以及(D) 还显示了配体结合的PKCδC1B结构域。 在面板B、C、, D、碱性、酸性和中性极性残基显示为蓝色, 红色和绿色。
尽管序列同源性和结构相似, 4 , 9 不同蛋白质的C1结构域表现出广泛的结合 对阴离子脂质辅活化剂如1,2的亲和力- 锡 -磷脂酰- 我 -丝氨酸(PS)以及中性脂质 类似1,2的活化剂- 锡 -二酰甘油(DAG)和佛波醇-12-氨基乙酸-13-醋酸盐 (PMA)。 特别是,PKCs的许多典型C1结构域结合DAG或 优先考虑PMA,而其他人则与两者具有类似的亲和力 激活器。 13 也存在非典型 既不绑定DAG也不绑定PMA的C1域。 4 这些在结合亲和力和偏好上的差异具有挑战性 以当前可用的有限结构为基础进行解释 数据本身。 目前仅沉积了13个不同的C1域 到蛋白质数据库(PDB),因此没有三维数据 90%以上具有已知序列的C1域的(3D)信息。 1 此外,复杂结构中只有一个结构 用X射线晶体学解决了任何脂质活化剂的问题。 11 因此,C1域的关键结构细节, 例如,与阴离子脂质和DAG以及 蛋白质的取向和膜的渗透程度,没有 已被充分阐明。 考虑到学习的实验挑战 膜结合环境中的C1结构域, 2 , 14 , 15 计算机建模与原子模拟 为探索详细的相互作用和构象提供了一种有用的方法 参与脂质激活剂的膜结合和识别, 这可能会增强我们对C1结构域作用的理解 用于信号蛋白的功能和调节。
提出了一种计算和实验相结合的研究 在里面 这项工作的目的是调查 PKCα亚型(PKCα)中的C1结构域及其脂质伴侣 (PS、DAG和PMA)。 牵涉到大量人类疾病 16 , 17 包括癌症, 18 , 19 心血管疾病, 20 , 21 糖尿病和并发症, 22 , 23 以及双极 混乱, 24 , 25 PKCα作为模型被广泛研究 对于传统PKC(cPKCs:α、βI、βII和γ 异构体),以了解C1结构域如何调节信号蛋白。 9 与其他cPKC类似,PKCα具有 两个串联C1域, 26 即C1A和 C1B,以及C2靶向结构域和C末端激酶 域。 9 证据表明 紧致非活性状态下的PKCα通过顺序激活 单个结构域与质膜表面的结合。 27 − 30 首先,钙触发C2结构域与阴离子脂质的结合 整个PKCα蛋白被导向膜。 正在解除关联 在激酶结构域中,抑制性C1A和C1B结构域是 吸收到膜上结合脂质辅活化物(如PS) 和活化剂(如DAG和PMA)。 此过程将激活 PKCα,催化底物蛋白的磷酸化。 9
最近取得了重大进展 29 , 31 强调重要性 激活机制中的两个C1结构域,以及 需要进一步的计算和实验努力才能更好地 了解这些结构域的膜相互作用。 成长中的身体 实验证据表明cPKC中的两个C1结构域 不是等效的,而是这种不等效的分子基础 只是部分理解。 10 例如, 尽管两个C1结构域都被认为与膜相互作用 证据表明,两个cPKC C1结构域中只有一个能结合 添加到脂质活化剂中。 32 其他结果表明 在PKCα的情况下,这两个域都与激活子结合 28 , 29 但却有着相反的亲和力。 13 , 33 人们普遍相信 PKCαC1A结构域与DAG的亲和力高于 C1B结构域,而后者与PMA的亲和力较高。 14 , 33 , 34 关于等价物的突变 或离子残基,C1A结构域的影响更为显著 而在C1B结构域上PKCα膜的结合和活化。 35 − 37 最后,在cPKC激活机制中,最近的工作表明 C1A通过结合稳定主要的活化中间体 首先是膜,即使在没有激活脂质的情况下。 29 在这个模型中,C1B向膜的募集 激活脂质是激酶激活的关键步骤。 29 , 38 进一步阐明C1A和C1B结构域在PKCα中的作用 激活,了解其详细的分子是至关重要的 与膜的相互作用。 因此,我们将计算 以及本研究中描述相互作用的实验方法 具有不同脂质的PKCαC1A和C1B结构域 膜环境。
由于缺乏实验 确定的结构 以及cPKC C1结构域的膜对接几何,这是一个挑战 利用计算机模拟比较 膜结合环境中的cPKC相同。 而大多数之前的对接 以及针对PKCα的C1B结构域和 溶液中的PKCδ, 37 , 39 − 41 在脂质双层中仅模拟了PKCγC1B结构域。 42 据我们所知 对膜中C1A和C1B结构域的研究从未报道过。 鉴于我们最近在PKCα方面的计算和实验进展 研究, 12 , 29 , 43 现在有了 采用组合方法进行揭示和比较变得可行 膜中两个PKCαC1结构域的细节。 如所示 在这项工作中,我们建立了原子PKCαC1A和C1B模型 在不同的膜结合环境中 结合模拟和实验结果的调查。 模拟和实验之间的协同作用使我们能够访问结构 C1结构域-脂质复合物在不同空间的动力学细节 和时间尺度。 我们的联合研究旨在帮助理解 PKCαC1结构域-脂质相互作用的原子细节 并提供有价值的新 C1结构域在PKCα调控中的作用 和激活。
结果和讨论 中的PKCαC1结构域 PC:PS膜 自PS以来 脂质是cPKC与细胞膜结合的重要辅活化剂, 44 我们首先进行了原子分子动力学 在3:1 PC:PS中使用单个C1A或C1B域进行的(MD)模拟 膜,以检查蛋白质和PS之间的相互作用。 在这两种情况下,C α RMSD在前20–26年上升 ns,然后在剩下的时间内稳定在1.5到2.0°之间 模拟结果证明了我们膜结合的结构稳定性 蛋白质模型。 与基准PKCα的主要偏差 溶液中的C1B结构(PDB代码:2ELI),是指 β12和β34环路尖端,定义为C α C1A域或S111和L125中Q46和F60之间的距离 在C1B域中。 37 增加的循环 分离导致激活剂结合槽的打开,尽管 在这两个域中的不同程度(请参见 支持信息(SI),图S1A ). C1B域显示 循环时间距离从11.5º略微增加到 平均值为12.2Ω,分布范围从9到 16Å。 在我们的模拟中采样的构象非常一致 观察到的开环构象(~12.5°) 在早期的模拟研究中使用相同的C1B域。 37
与C1B域相比,C1A域 似乎具有更宽的激活剂结合槽,可能是由于 存在更灵活的β12和β34环。 这个 相比之下,环距通常要长2–3奥 到C1B域的相应距离,具有分布 以15.3°为中心( 图S1A(SI) ). 总的来说,在缺少活化剂的情况下,分离度增加 循环尖端以及由此产生的开放激活器绑定槽 在PKCαC1结构域中,表明与PC:PS的相互作用 膜稳定开环构象并可能促进 激活剂相互作用。
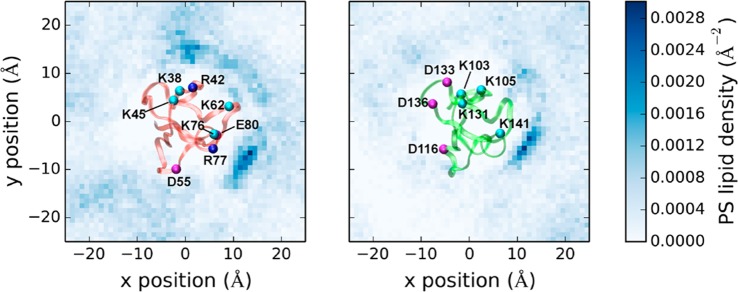
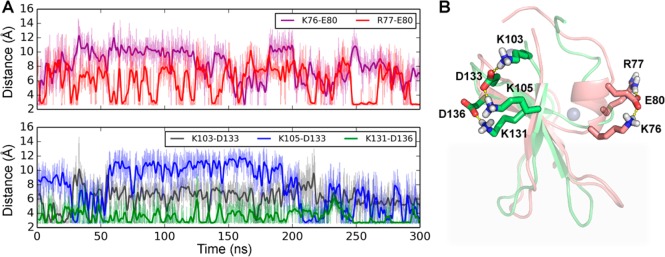
与PC:PS膜诱导 构象的 C1结构域的改变,蛋白质也形成了局部PS分布。 PS头部组的横向分布如图所示 2 揭示了两者的异同 C1A和C1B域之间的相互作用 PS脂类。 一方面,由于在蛋白质中没有检测到密度 区域,PS头组仅在 C1结构域,不包括在激活剂结合凹槽中 在这两种情况下。 很可能,在他们作为共同激活者的角色中, 阴离子脂质(如PS)几乎不干扰活化剂结合。 另一方面,如不同的高密度区域所示, PS脂质在非常不同的位置与两个C1结构域相互作用。 PKCαC1A结构域具有许多基本残基(即R42, K45、K62、K76和R77),对应于半环形 PS密度高的区域。 与低密度相邻的另一面 该区域主要由几个芳香族残基(即F49、, F56、W58和F72)和酸性残留物D55。 相反,PKCα C1B结构域仅在两个基本残基K105附近具有高PS密度 和K141。 C1B结构域周围的大部分膜区显示 PS密度远低于C1A域外围观察到的PS密度。 这一现象不能简单地归因于基础知识少而基础知识多 PKCαC1B域中的酸性残基比C1A域中的酸残基多。 我们对离子残基的进一步分析揭示了一个稳定的盐桥 C1B域中的网络,这导致以下解释: 两组持续相互作用的带电残基K103-D133-K105 和K131-D136,在C1B域中形成盐桥网络(图 三 ). 这样的盐桥网络(i)减少了数量 和(ii)削弱蛋白质与PS的接触,因此 C1B结构域吸引PS脂质的能力较差。 相比之下, 在C1A域中未观察到类似的网络: 残留物K76-E80-R77之间的静电接触不同,但 基本残留物仍然可以结合PS脂质(图 三 和 4 ). 因此,我们的模拟 表明不同的静电详细相互作用 观察到PKCαC1A和C1B的不同结合 具有PS脂质的结构域。
图2。
PKCα的PS头组密度图 C1A域(左侧面板) 和C1B域(右侧面板)。 蓝色轮廓显示表面 PS脂类头部群密度以下膜小叶为主 在300ns模拟中。 从底部观察 模拟框中,蛋白质主干显示为色带 C α 离子残留物的原子成珠状。
图3。
(A) 分子内盐桥的时间演化 在300纳秒 PKCαC1A(顶部面板)和C1B域(底部面板)的模拟 面板)中:PS膜。 对原始数据和平滑数据进行了说明 分别为细线和粗线。 (B) 结构表示法 在载脂蛋白C1A模拟的290 ns处盐桥网络(粉红色 主干)和载脂蛋白C1B模拟293 ns处(绿色主干)。 锌 2+ 离子显示为银球。 为了说明 薄膜插入,阴影框用于指示薄膜 作为眼睛的向导。 配体结合位点在两者中都是开放的 构象。
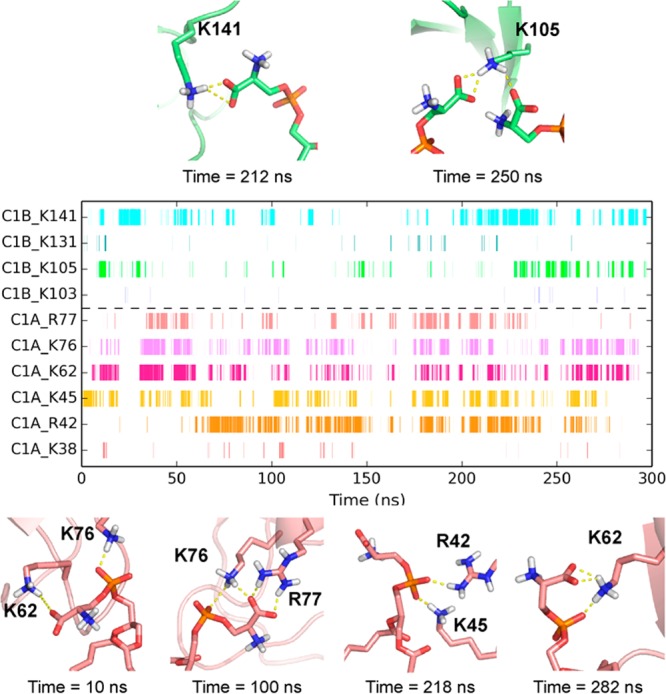
图4。
事件图 与PS相关的关键基本残基 脂质。 事件图中的每一行表示 含有结合PS脂质的碱性残渣。 不同位置的模拟快照 以时间间隔表示多价结合。
即使是短暂的 在我们的模拟中,PKCαC1A结构域 可以结合多达4–5个PS脂质,而PKCαC1B 该结构域可以结合多达2–3个PS脂质(图 4 ),PS结合化学计量比(时间平均值)仅为1.2 分别为±1.0和0.3±0.6。 巨大的不确定性 这些值突出了C1域-PS关联的动态性质。
在这些MD研究之后,进一步的证据 与…一致 C1A结构域的较高正电荷和PS化学计量比 相对于C1B结构域,通过单 分子TIRF显微镜(SM-TIRFM)研究。 如图所示 5 ,这些实验表明 C1A结构域的结合亲和力对PS更敏感 密度高于C1B域。 正如预期的那样,这两种蛋白质都结合双层 当PS脂质 存在。 有趣的是,我们的SM-TIRFM实验发现C1A 结构域对含PS的膜具有明显更高的亲和力 而不是C1B结构域,这一发现遵循了其相对 MD模拟建议的PS结合化学计量。
图5。
依赖 磷脂酰丝氨酸上的C1A和C1B双层结合 (PS)密度。 通过SM-TIRFM定量蛋白质的膜密度 如前所述。 29 C1A或C1B浓度 固定在5 pM,并添加到支撑磷脂酰胆碱(PC)中 含有(A)增加PS脂质量或(B)增加的双层膜 PS脂质总量,其中阴离子脂质总量保持在40%,使用 磷脂酰甘油(PG)脂质。 短暂孵化后,确保 荧光膜结合蛋白的稳态结合密度 从三个单独的框架中测量了5个暂时隔离的框架 电影流在三个单独的滴定实验中。 为了 去除PG脂质引起的纯静电结合 去除0~40%PS脂质的结合信号。 产生的结果 因此,结合是由于与PS的特定蛋白质相互作用,因为 非特异性静电征募已被消除。
更详细地检查中的PS绑定事件 我们的模拟 表明没有一个已识别的相互作用位点持续存在 绑定到PS(图 4 ). 例如,每个 PKCαC1A结构域中的残基R42、K45、K62和K76 26-30%的时间与PS脂质相关 17%的时间使用R77,而K105和K141位于C1B域 显示仅约10%和18%与PS脂质相关 时间。 这些结果表明C1域-PS关联 不饱和,因此亲和力不高。 然而,如图所示 通过我们模拟的快照(图 4 ),至少一些PS结合位点提供多个头组 联络。 给定PKCαC1结构域中的基本残基 PS对映体(1,2- 锡 -磷脂酰- 我 -丝氨酸) 似乎非常适合结合多个基本残基, 可能是由于两个分离的负电荷中心 以及立体化学。 我们估计 在我们的模拟中,C1-PS多价触点更好地通过以下方式实现 生理对映体1,2- 锡 -磷脂酰- 我 -丝氨酸比非生理对映体2,3- 锡 -磷脂酰- d日 -丝氨酸。 这与 牛顿及其同事的发现表明 对映体2,3- 锡 -磷脂酰- d日 -丝氨酸 对PKC的亲和力显著降低。 45 此外,我们的模拟也提供了结构证据 支持早期实验 45 , 46 他们发现了 cPKC对PS的亲和力高于其他脂质(如PC、PA、, 和PE)。 因此,尽管没有高亲和力的PS结合位点 在PKCαC1结构域中观察到多价C1-PS相互作用 可以解释所观察到的C1对PS对映体的立体特异性。
总之,我们的MD模拟和SM-TIRFM的发现 实验表明,正合作性的一个简单机制 在实验C1结构域结合测量中观察到 PS密度变化,并保持固定的双层负电荷 (图 5 ). 在这个模型中,C1域首先 通过与单个PS分子相互作用与双层结合, 然后与一个或多个额外的PS分子相互作用。 在这样的系统中,初始PS关联将增加亲和力 导致积极合作。
PC:PS膜中含有DAG/PMA活化剂的PKCαC1结构域 为了探索C1域激活子的详细相互作用,我们模拟了 PC:PS膜中活化剂结合的C1复合物 未绑定的激活器。 在缺乏配体结合的结构信息的情况下 PKCαC1结构域,我们测试了不同的激活物结合姿势和 对于每个复杂体,只获得一个对整个复杂体稳定的姿势 300 ns膜结合模拟,主要使用蛋白CαRMSD 波动范围为1.0至2.6º。 与无激活剂相比 例,DAG结合诱导的平均1.8和1.6μ C1A和C1B分别在环距中,而PMA绑定 在所有配体结合的情况下,诱导的3.0和0.5 Au分别降低 产生更窄分布的状态(参见 图S1(SI) ). 这些观察结果表明激活剂 结合导致凹槽闭合,蛋白质含量更高 刚性。
PS与PKCαC1结构域的关联不是 改变了的 在我们的模拟中,脂肪活化剂的存在显著。 据估计 带DAG的C1A和C1B域分别为1.0±1.0和0.2±0.4 PMA为0.9±1.0和0.4±0.6,表明 C1结构域和PS的相互作用仍然是高度动态的。 专业 PS相互作用的位点与无激活物的情况相同: C1A域中的R42、K45、K62、K76和R77,以及中的K105和K141 C1B域。 这些结果再次支持绑定的概念 C1结构域对脂质活化剂的直接影响很小 它们与PS脂质的关系。
为了进一步检查 为什么PKCαC1A和C1B结构域 可能有相反的亲和力来结合DAG和PMA, 33 我们比较了氢键和非极性接触 所有四个复合物。 令人惊讶的是,一个高度保守的氢键 观察到了模式,尽管此模式似乎不够 解释两人对激活脂质的不同偏好 域。 保守残基的主干,包括T48、I57和 C1A域的G59和C1B域的T113、L122和G124, 与DAG和PMA形成一致的氢键(图 6 ). DAG的1-羟基或PMA的20-羟基,用于 作为氢键的供体和受体,连接着主链 C1A结构域中的I57和T48原子或C1B中的L122和T113原子 域(图 6 C1、C2)。 I57/L22主干 NH向T48/T113主链羰基提供氢键,因此 桥接β12和β34环(图 6 C1、C2)。 这个氢键三角形很可能决定了 激活器在绑定凹槽中的定向。 此外,DAG和 PMA还与G59和G124的主链原子形成氢键 进一步锚定复杂构象。 在没有水的情况下, 这种氢键网络对激活剂的访问起着关键作用 结合槽,以及稳定其C1结构域 膜结合态。 值得注意的是,Raf-1 C1域, 47 与DAG缺乏结合的非典型C1结构域, 由于短得多,不能形成这些氢键 β34环(参见 图S2(SI) ),其中 强调了这些特殊氢键的重要性。
图6。
(A1)叠加DAG-bound C1A和 C1B构象。 构象是从代表处获得的 最可能的复杂集群。 卡通插图(A2,3) DAG和C1A结构域之间的氢键,以及(A4,5)氢 DAG和C1B结构域之间的键。 (B1)结构表示 叠加PMA结合的C1A和C1B构象。 (B2,3)氢 PMA与C1A和C1B结构域之间的键。 (C1,2)插图 激活剂和蛋白质之间的氢键。 (D) 非极性接触的比较。
而涉及蛋白质骨架的氢键网络 似乎在PKCαC1和PKCδC1B之间保守 域, 11 侧链极性接触 显示为DAG和PMA之间的明显差异。 在我们的模拟中, C1A结构域Q63和C1B结构域Q128的侧链 氢键不仅与β12和 β34环路,但也有DAG(图 6 A3、A5)。 然而,在Q63/Q128和 PMA,因为在这种情况下谷氨酰胺侧链的拉伸较小 无法提供联系人。 我们的结果进一步表明, 支持关于Q128的早期发现的一般证据 PKCαC1B结构域, 37 这表明 这种保守的谷氨酰胺残基在调节 活化剂结合槽的形状和接触。
虽然守恒的氢键网络仍然不能解释 PKCαC1A和 C1B域,我们对非极性接触的分析确实提供了一个可信的 对这种差异的解释。 在图中 6 D、, 激活器和C1之间的非极性接触数量 根据定义中不断增加的截止值绘制域 接触距离。 DAG总是有更多的非极性接触 与C1B域相比,C1A域不考虑截止值 被雇佣。 PMA观察到相反的行为 与C1B结构域有更多非极性接触。 从更改时 DAG到PMA,C1A的非极性触点数量减少,但 C1B增加。 PISA界面能估算 48 表明C1A-DAG复合物更稳定 C1A-PMA络合物比C1A-PMA-络合物高2.1 kcal/mol,而C1B-PMA络合物 仅比C1B-DAG复合物稳定0.8 kcal/mol。这些结果 因此,为绑定偏好提供了半定性证据 C1域。 33 此外,我们的模拟 还能够识别最相关的残留物(2个以上 平均激活器触点),包括F43、P47、W58、F60、, 绑定到DAG时C1A域中的L63,以及P112、Y123、L125, 当绑定到PMA时,C1B域中的Q128。 这些发现是一致的 之前的诱变实验, 35 哪一个 表明其中一些残留物(尤其是W58和F60) 对DAG活化剂结合至关重要。
除DAG/PMA外 交互,我们还监视未绑定的 每个模拟中的配体(请参见 图S3(SI) ). 即使未结合的DAG或PMA分子足够接近 在我们的起始构象中C1域,它们不驻留 靠近蛋白质。 在300毫微秒模拟结束时,所有数据均未绑定 DAG和PMA配体远离蛋白质,表现出分离 距离最近的蛋白质重原子超过10μl。 符合 之前的实验观察, 12 我们的 模拟没有显示DAG的C1域中的次要站点 或PMA结合,表明活化剂的结合化学计量比为1:1 结合到每个PKCαC1结构域。
PKCα的运动 膜中的C1结构域 我们 未观察到膜结合蛋白激酶CαC1的明显倾斜 模拟中的域,如小值和窄值所示 最长蛋白质主成分间夹角的分布 轴和 Z轴 -轴。 这些角度为17±8 对于C1A域和16±8度的C1B域 PC:PS膜,C1A达到16±8 PC:PS+DAG膜中C1B结构域为12±7度 以及PC:PS+PMA中的14±8(C1A)和13±9(C1B)度。 这些结果表明PKCαC1结构域的取向一致 插入膜中,再次确认整体稳定性 在我们的模拟中使用的模型。
此外,我们检查了 PKCαC1结构域的膜透性。 我们的模拟 表明这两个结构域在膜中没有固定。 定义 作为域重心到DOPC N平面的距离, C1A和C1B结构域的膜插入深度可以连续变化 从深部状态(~−2º)到浅部状态 (~10º),分布如 图S4(SI) 在深态下 C1结构域嵌入膜中,而处于浅态 线圈的尖端几乎不接触薄膜。 这些结果证实, 在分子水平上,膜结合PKCα的两种状态 我们早期实验中发现的C1结构域, 29 虽然MD模拟可能不够长,无法完全 定义在深层和浅层状态中花费的时间比率。 这个 所有薄膜的平均插入深度为3.9–4.0º 我们的构造,但PMA-bound C1B域除外,该域具有平均值 4.4º的值(参见 图S4(SI) ). 建议DAG或PMA绑定通常不会修改 C1结构域的膜渗透,除了PMA诱导 C1B域移动到更浅的位置。
除了 定向和膜渗透,我们有 还通过模拟测量了蛋白质扩散系数 就像实验一样。 虽然在膜中进行这样的计算非常困难 我们注意到,为了收敛,应该谨慎看待其结果 我们的原子模拟与早期相比相当好 独立脂质双层中的粗颗粒膜蛋白。 15 , 49 虽然原子论的绝对扩散系数 模拟值比实验值大大约一个数量级 值,我们在模拟和实验之间实现了定性一致 (表 1 ). 尽管膜不同 构造(用独立双层模拟与实验 模拟和实验都同意 PKCαC1结构域具有非常相似的扩散系数 ( D类 L(左) )在含有或不含活化剂的膜中, 除了PMA结合的C1B域具有更高的 D类 L(左) 而不是其他组件。 正如之前工作中建议的那样, 12 , 29 膜透性是膜蛋白的重要决定因素 周边扩散。 很可能浅插入深度 在我们的模拟中观察到的PMA-bound C1B域与 在两个模拟中测量的增加的扩散系数 和实验。 PMA-C1B仿真的独特发现 复杂的是,通过 150至300毫微秒,约2.8欧姆。 事实上 插入深度小于4.4º的构象增加 仅PMA-bound C1B结构域每50 ns约20%,而 其他结构未见明显增加。 此外, PMA-bound C1B域独有的激活器尾对尾触点 到PC或PS的脂质从~200到300毫微秒减少50% 在我们的长模拟中。 因此,我们假设联合效应 与PS头组的弱相互作用和接触减少 到膜疏水核心导致更高的 浅层状态,可能导致 PMA结合的C1B结构域的快速扩散。 相对较大的 PMA对C1B实验扩散系数的影响 源于扩散的较长时间尺度和空间维度 测量。
表1。 外围扩散系数 D类 L(左) 从独立式MD仿真中获得 双层膜和支撑双层膜的实验(单位:μm 2 /s) 一 .
PKCα C1A公司 PKCαC1B
仿真 实验 仿真 实验
个人电脑:PS 12.8 ± 1.8 0.7 ± 0.1 15.1 ± 1.4 0.5 ± 0.1
个人电脑:PS+DAG 14.6 ± 2.2 0.7 ± 0.1 15.6 ± 2.1 0.6 ± 0.2
个人电脑:PS+PMA 12.3 ± 1.8 0.7 ± 0.1 20.3±3.0 1.2±0.1
详细的膜相互作用机制 PKCαC1 域 将我们系统组合的证据联系起来 研究中,我们提出了可能的详细膜结合机制 PKCαC1结构域。 与PKCαC2结构域相比, 我们之前的工作已经研究过了, 43 我们发现PS与C1的相互作用弱于与 C2.根据之前提出的激活机制, 29 C2域而不是C1域 可能将PKCα导向膜。 对于后续 活化步骤,尽管这两个C1结构域的溶剂暴露 在全长PKC可能不同的情况下,我们的研究支持这一观点 C1A结构域在C1B结构域之前被膜吸收 因为它与阴离子PS脂质的相互作用更强。 一次 它与膜结合,C1A或C1B结构域可能经历 构象变化打开激活物结合槽,而 同时,整个区域在浅层之间波动 和深层膜插入状态。 深层膜插入增强 开口槽构象的稳定性,这可能与 激活器的搜索机制。 当PKCαC1结构域 绑定激活器,激活器绑定槽可能会关闭, 整个领域变得更加僵化。 保守氢键 对激活物识别和结合方向很重要 膜结合环境。 然而,它是非极性接触 C1域和导致相反结果的激活子之间 活化剂结合偏好。 最后,PMA结合似乎有利于 C1B结构域的浅结合状态,如MD中观察到的 模拟和实验扩散系数建议。 PKCαC1B和 PMA,膜和C1B-PMA复合物之间的接触减少, PMA结合的C1B结构域的异常快速扩散可能与此有关 PMA等诱导肿瘤发生的分子机制 佛波酯。
结论 我们有组合建模, 模拟和实验研究 PKCα在细胞膜中的C1A和C1B结构域是一项艰巨的任务 只有一种方法。 我们以前的发现 12 , 29 , 43 目前的工作表明 以下详细机制涉及C1A和C1B域 PKCα激活:在整个PKCα与 膜,两个C1结构域都可以在激活期间与膜结合。 C1A结构域首先通过与脂质的强相互作用被募集 共激活子PS和激活子DAG,C1B结构域被招募 稍后,首选绑定激活器PMA。 两个PKCα C1域在其序列中编码以发挥不同的作用, 通过与助凝剂的不同表面静电接触 以及与激活剂的非极性接触。 我们的研究提供了 支持C1B通过以下方式与膜结合的观点的证据 活化脂质可能是PKCα活化的关键步骤 模型。
此外,确凿证据来自 仿真 以及脂质结合和蛋白质扩散方面的实验。 仿真 实验相辅相成,使我们能够联系证据 C1域相互作用的多个时空尺度 带有膜。 我们的组合方法将有助于探索 C1结构域在许多信号蛋白中的作用,甚至在 缺乏详细的结构信息,有助于进一步了解 它们在正常细胞功能和疾病中的分子机制 发展。 考虑到这里使用的原子论MD模拟方法, 重要的是要意识到我们模拟之间的差异 以及在时间和长度尺度上的实验。 未来的努力将是 开发精确的粗颗粒脂质和蛋白质模型 在实验时间尺度上解释和预测蛋白质动力学。 为了获得关于PKCα激活的更多信息,我们 还利用所获得的知识模拟了一个全长模型 来自我们PKCα研究中的各个领域 12 , 29 , 43 以及这项工作。
材料和方法 膜结合模型构建 我们已经建模并 模拟了膜和缓冲液中的单个C1A和C1B区域 模拟实验条件。 在许多实验中 C1结构域与~300-残留麦芽糖结合蛋白融合 (MBP),用于增强C1结构域的溶解度。 29 如所示 支持的 问询处 ,我们确认MBP和肽 连接器对C1之间的相互作用影响可以忽略不计 域和膜(参见 图S5(SI) ). 因此,只对单个C1域建模是合理的 为了了解详细的蛋白质-脂质相互作用。
最初,建立了膜模型和蛋白质模型 独立地。 3:1 PC:PS对称双层模型,包含120个 DOPC和40种DOPS脂质,使用CHARMM-GUI膜构建器进行设置 50 并在水箱中预平衡(包含 150 mM NaCl溶液),使用CHARMM36力场和 Desmond 3.0仿真包。 51 这个 然后将膜模型与 X – Y(Y) 平面,垂直于 Z轴 -轴。 这个 PKCαC1B模型(残差102–151)基于坐标 残基18-67的溶液结构(PDB代码:2ELI)。 PKCα C1A模型(剩余37–86,图 1 ) 由同源建模服务器SWISS-MODEL生成, 52 与模板(PDB)具有42%的序列一致性 代码:1PTR)。 为了检测离子残基的质子化状态,蛋白质 在Maestro中实现的准备向导(版本9.3,Schrödinger, LLC,2012年)。 每个锌 2+ 离子被三个 硫代形式的半胱氨酸残基和一个组氨酸残基 在我们开始的蛋白质构象中,质子化在δ-氮原子上。
然后将PKCαC1A和C1B模型与PC:PS相结合 膜。 对每个C1A或C1B结构进行对齐、旋转和移动 在膜模型下方,因此蛋白质的长轴是 几乎与 Z轴 -轴和尖端 β12和β34环可以指向下叶 膜的厚度。 根据初步实验数据, 12 , 29 蛋白质中心位于DOPC的N平面以下6° 将下部小叶插入蛋白模型到膜模型中。 几乎一半的C1A/C1B结构域位于下叶,但确实如此 不能到达上小叶。 使用Maestro中的System Builder 在每个构造中创建包含140 mM NaCl和10 mM KCl的盒子, 其边界与最近的蛋白质或 Z方向的脂原子。 因为在C1之间存水 域和膜通常是不利的, 11 水分子被排除在蛋白质膜附近 接口。
将DAG和PMA分子插入膜中, 为了 在有激活物的情况下对C1域建模。 PMA绑定 根据配体结合PKCδ的排列建立模型 C1B结构(PDB代码:1PTR)到上述模型中的蛋白质结构, 然后将佛波酯配体改性为PMA。 自 已发现DAG与PMA竞争相同的结合位点, 53 我们将DAG的选定氧原子与 晶体结构中的佛波醇化合物,因此 在我们的搜索中构建并测试了DAG-bound C1A和C1B构造 用于稳定络合物。 除了结合活化剂,还有两个DAG 或将PMA分子插入蛋白质附近进行检查 任何二级结合或相互作用位点。
简言之,三 膜成分(PC:PS,PC:PS+DAG,PC:PS+PMA) 对于PKC,使用αC1A和C1B结构域构建6个结构体。 每个构造包含大约52个 a~85中有000个原子 周期性边界的Φ×85Φ×85Ω盒 条件。
原子MD模拟 我们申请了 工具Viparr 在Desmond中指定全原子力场参数。 蛋白质 参数是从CHARMM27 cmap力场获得的,除了 硫代参数采用了先前的报告。 54 CHARMM36参数用于DOPC和 DOPS以及DAG和PMA的CHARMM通用力场。 55 TIP3P模型用于显式水 分子。 参数赋值后,初始模型最小化 以消除立体碰撞,并使用中的标准方案放松 Maestro-Desmond套餐。 我们在包中使用了一个脚本 M-SHAKE算法约束涉及的所有键的键长 氢原子,以及所有水分子的角度。 我们的产品 用Desmond 3.0进行了模拟,时间步长为2 fs。 键合和近相互作用每一步都会更新,而 far交互每三步更新一次。 这些半同位素 模拟是在恒定温度(296 K)和恒定温度下进行的 压力(1巴)。 Nosé–Hoover链恒温器方法 与Martyna–Tobias–Klein一起受雇 气压调节器。 计算库仑和伦纳德-琼斯的短程截止值 相互作用为9.0℃。 长程库仑相互作用 采用光滑粒子网格Ewald方法进行处理。 两个副本 对每个构造进行了模拟:一个短构造为150ns 一个长的300纳秒。 我们的短期和长期模拟的一致性 表明我们的模拟时间范围足以消除启动 构象偏差。
仿真数据分析 构象的 分析是 使用VMD执行 56 和Pymol(Schrödinger, 有限责任公司)。 在这项工作中,蛋白质的根平方偏差(RMSDs) 构象在C上计算 α 原子对 与参考PKCαC1B结构对齐(残基18-67, PDB代码:2ELI)。 盐桥或带电接触是指两个极性原子 相反电荷在4.0°以内。 结合脂质的定义 作为那些在距离最近的蛋白质4.0Å以内有重原子的蛋白质 带有相反电荷的重原子。定义了疏水接触 当两个非极性原子(部分电荷<0.3单位)位于 截止距离。 测量膜中的蛋白质取向 作为蛋白质最长主轴与 这个 Z轴 -轴。
周边扩散系数 ( D类 L(左) )PKCαC1结构域的 根据随时间变化的均方位移(MSD)计算 至eq 1 :
哪里 第页 是重心 C1A或C1B域的向量。 计算平均值 分割模拟中的块。 下部的整体漂移 从蛋白质扩散中去除传单, 57 而在 我们的MSD计算。 MSD与时间的关系图如所示 图S6(SI) .
最长之间的角度 蛋白质主轴和 Z轴 -轴已测量 检查蛋白质的取向 膜。 为了量化膜渗透,膜插入 PKCαC1结构域的深度定义为 蛋白质的质心到最近的氮平面 DOPC脂质。
实验 试剂和实验 协议是 与之前描述的工作一致 12 , 15 , 29 并将进行简要描述。
细菌 人PKCαC1A和C1B调节域的表达结构 通过插入编码C1A结构域的DNA序列(残基 26–100)和C1B结构域(90–165)转化为pMAL-c2G表达 矢量。 针对每种蛋白质,设计引物以结合 Sfp磷酸-对噻吩基转移酶的N-末端11-氨基酸识别序列 以使得能够用CoA连接的荧光团进行序列特异性酶标记。
C1A和C1B结构域在 大肠杆菌 Rosetta 2(DE3)细胞(Novagen)。 20°C下过夜表达 然后在直链淀粉树脂(NEB)上纯化,并用 过量麦芽糖。 纯化蛋白≥总洗脱蛋白的90% 蛋白质。 N末端Sfp标签被共价修饰 通过Sfp酶合成Alexa Fluor 555-CoA,过量荧光团 使用Vivaspin集中器(Sartorius Stedim,哥廷根, 德国)。
要生成支持的双层,需要使用声波单层 囊泡 (SUV)由合成二油醇磷脂PC(磷脂酰胆碱; 1,2-二油酰- 锡 -甘油-3-磷酸胆碱)、PS(磷脂酰丝氨酸; 1,2-二油酰- 锡 -甘油-3-磷酸- 我 -丝氨酸), DAG(二酰甘油;1,2-二油酰基- 锡 -甘油),PG (1,2-二油酰基- 锡 -甘油-3-磷酸-(1′- rac公司 -甘油)[均来自Avanti Polar Lipids(雪花石膏, AL)]和PMA(佛波醇-12-嘧啶酸13-乙酸酯)[来自Sigma-Aldrich (密苏里州圣路易斯市)]沉积在食人鱼清洁的玻璃基板上。
TIRF显微镜测量在22°C± 如前所述,在自制客观仪器上为0.5°C 描述。 12 , 15 , 29 在添加 生理缓冲液(140 mM KCl、15 mM NaCl、0.5 mM MgCl、26μM 氯化钙 2 ,20μM EGTA,减少5 mM 我 -谷胱甘肽, 25 mM HEPES,pH 7.4)和BSA阻断步骤 荧光污染物通常很少。 5分之后 与蛋白质孵育min,样品在高激光功率下漂白 将不动荧光粒子的贡献降到最低 通过60秒的恢复。 对于每个样本,采集了多个电影流 帧速率为20帧/s,空间分辨率为4.2像素/μm。 使用ImageJ进行粒子跟踪分析和拟合, GraphPad Prism 5和Mathematica。
致谢 这项研究得到了支持 美国国立卫生研究院 授予R01 GM-063796(给G.A.V.)和R01 GM-0.63235(给J.J.F.)。 这个 使用极限科学与工程发现环境进行的工作 (XSEDE),由美国国家科学基金会(National Science Foundation Grant Number)资助 OCI-1053575。 计算资源由XSEDE Kraken提供 以及芝加哥大学的研究计算中心(RCC)。 我们感谢Anand Srivastava博士和Severin T.Schneebeli博士的帮助 讨论。
可用的支持信息 环距分布 用原子论MD模拟(图S1)测量叠加结构 不典型C1结构域和PKCαC1结构域的DAG-bound模型 (图S2),未结合活化剂的横向扩散路径(图 S3)、膜插入深度的比较(图S4)和分析 MBP-C1A-C1B仿真的最终快照(图S5)。 均方根 位移与时间曲线(图S6)。 此材料可用 通过互联网免费访问 http://pubs.acs.org .
工具书类
赫尔利·J·H。; 米斯拉S。 每年。 生物物理版。 生物摩尔。 结构。 2000, 29, 49. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
赫尔利·J·H。 生物化学。 生物物理学。 分子细胞生物学学报。 脂质 2006, 1761, 805. [ 谷歌学者 ]
赵伟(Cho W.)。; Stahelin R.V.蛋白质-脂质 相互作用; Wiley-VCH Verlag股份有限公司& KGaA公司:Weinheim,2006年; 第367页。 [ 谷歌学者 ]
赫尔利·J·H。; 牛顿A.C。; 帕克·P·J。; 布隆伯格下午。; 西冢Y。 蛋白质科学。 1997, 6, 477. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
梅勒·H。; 帕克·P·J。 生物化学。 J。 1998, 332, 281. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Cho W.H。; 斯塔赫林R.V。 每年。 生物物理版。 生物模型。 结构。 2005, 34, 119. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
罗斯·C。; 林奇·M。; 科尔摩根公司。; 卡梅隆·A·J。; Boeckeler K。; 帕克·P·J。 自然修订版分子细胞生物学。 2010, 11, 103. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
巴尔丹齐G。 高级生物。 雷古尔。 2014, 55, 39. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
斯坦伯格公司。 生理学。 版次。 2008年,881341。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
科隆-冈萨雷斯F。; Kazanietz M.G.公司。 生物化学。 生物物理学。 学报 2006, 1761, 827. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
张国国。; Kazanietz M.G。; 布隆伯格下午。; 赫尔利·J·H。 单元格 1995, 81, 917. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
齐安巴B。 体育。; 福克·J·J·。 化学。 物理学。 脂质 2013, 172–173, 67. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
斯莱特·S·J。; Ho C。; Kelly M.B。; 拉金·J·D。; 塔迪奥·F·J。; Yeager医学博士。; 斯塔布斯·C.D。 生物学杂志。 化学。 1996, 271, 4627. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Shindo M。; 艾丽·K。; Nakahara A。; Ohigashi H。; Konishi H。; Kikkawa U。; 福田H。; 温德尔P.A。 生物有机医药化学。 2001, 9, 2073. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
奈特·J·D。; 勒纳M.G。; 马卡诺·弗拉兹奎兹(Marcano-Velazquez J.)。 G。; 牧师R.W。; 福克·J·J·。 生物物理学。 J。 2010, 99, 2879. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Konopatskaya O。; 普尔A.W。 趋势药理学。 科学。 2010年,31日,8日。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
莫奇利·罗森D。; 达斯·K。; 格里姆斯公司。 Nat.Rev.药物发现 2012, 11, 937. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
格里纳·E·M。; Kazanietz M.G.公司。 Nat.Rev.癌症 2007, 7, 281. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Kim J。; 索恩·S·H。; 孙磊。; 黄B。; 莫奇利·罗森D。 癌基因 2011, 30, 323. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
莫尔肯廷J。 循环 2008年,第118页,第18页。 [ 谷歌学者 ]
刘庆华。; Molkentin J.博士。 分子细胞杂志。 心脏病。 2011, 51, 474. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
杰拉尔德斯·P。; 国王G.L。 循环。 物件。 2010, 106, 1319. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
凯勒M。; 穆沙克J。; 塞弗·E。; 米沙克H。; Ullrich A。; 哈林H.U。 糖尿病学 1998, 41, 833. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
萨拉特C.A。; Manji香港。 中枢神经系统药物 2009, 23, 569. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Manji香港。; Lenox R.H.公司。 临床杂志。 精神科 2000, 61, 42. [ 公共医学 ] [ 谷歌学者 ]
伯恩斯·D·J。; 贝尔R.M。 生物学杂志。 化学。 1991年、266年、18330年。 [ 公共医学 ] [ 谷歌学者 ]
牛顿A.C。 生物学杂志。 化学。 1995, 270, 28495. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
伦纳德·T·A。; Rozycki B。; 赛迪L.F。; 悍马G。; 赫尔利·J·H。 单元格 2011, 144, 55. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Ziemba B.P。; 李杰。; Landgraf K.E。; 奈特·J·D。; Voth G.A。; 福克·J·J·。 生物化学 2014, 53, 1697. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
斯科特·A.M。; 蚂蚁C.E。; 牛顿A.C。 生物学杂志。 化学。 2013, 288, 16905. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
佩雷兹·拉腊。; Egea-Jiménez A.L。; 奥西利A。; 科尔巴兰·加西亚。; 戈梅斯·费尔南德斯J.C。 生物化学。 生物物理学。 分子细胞生物学学报。 脂质 2012, 1821, 1434. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
乔治·J。; 海塞尔·M。; 哈维·D·F。; 牛顿A.C。 生物化学 2003, 42, 11194. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Ananthanaarayanan B。; 斯塔赫林R.V。; 迪格曼医学硕士。; 赵伟(Cho W.)。 J。 生物化学。 2003, 278, 46886. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Dries D.R。; 加列戈斯有限公司。; 牛顿A.C。 生物学杂志。 化学。 2007, 282, 826. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
梅德科娃M。; Cho W.H。 生物学杂志。 化学。 1999, 274, 19852. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Bittova L。; 斯塔赫林R.V。; 赵伟(Cho W.)。 生物学杂志。 化学。 2001, 276, 4218. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
斯图尔特医学博士。; 摩根银行。; Massi F。; 伊古梅诺娃T.I。 分子生物学杂志。 2011, 408, 949. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
蚂蚁C.E。; 小提琴J.D。; Kunkel M.T。; Skovso S.公司。; 牛顿A.C。 化学。 生物。 2014, 21, 459. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
木村K。; Mizutani M.Y。; 富冈N。; Endo Y。; Shudo K。; 意大利A。 化学。 药学公牛。 1999, 47, 1134. [ 谷歌学者 ]
Pak Y。; Enyedy I.J。; 瓦拉迪·J。; 龚建伟。; Lorenzo P.S。; 布隆伯格下午。; 王S.M。 医学化学杂志。 2001, 44, 1690. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
康建华。; 苯扎菌属。; 西格诺医学博士。; 勒温N.E。; Pu Y.M。; 桃子M.L。; 布隆伯格下午。; 马尔克斯V.E。 医学化学杂志。 2006, 49, 3185. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
赫里茨·J。; 尤利克尼·J。; Laaksonen A。; Jancura D。; 米斯科夫斯基P。 医学化学杂志。 2004, 47, 6547. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
赖C.-L。; Landgraf K.E。; Voth G.A。; 福克·J·J·。 分子生物学杂志。 2010, 402, 301. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
牛顿A.C。; 凯伦·L·M。 生物化学 1994, 33, 6651. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
约翰逊J.E。; 齐默尔曼M.L。; Daleke D.L。; 牛顿A.C。 生物化学 1998, 37, 12020. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
约翰逊J.E。; 乔治·J。; 牛顿A.C。 生物化学 2000, 39, 11360. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
莫特·H·R。; Carpenter J.W。; 钟S。; Ghosh S。; 贝尔·R·M。; 坎贝尔S.L。 程序。 国家。 阿卡德。 科学。 美国。 1996, 93, 8312. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Krissinel E。; Henrick K。 分子生物学杂志。 2007年,第372774页。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
鹅J.E。; 桑索姆M.S.P。 公共科学图书馆计算。 生物。 2013年9月,e1003033。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Jo S。; Lim J.B。; Klauda J.B。; 我是W。 生物物理学。 J。 2009, 97, 50. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
鲍尔斯·K·J。; 周瑜。; 徐宏。; Dror R.O。; 伊斯特伍德M.P。; 格雷格森B.A。; 克莱佩斯J.L。; 科洛舍利一世。; Moraes医学硕士。; 萨克多蒂基金会。; 三文鱼J.K。; Shan Y。; Shaw D.E.在2006年ACM/IEEE会议记录中 关于超级计算; ACM:佛罗里达州坦帕市,2006年; 第84页。 [ 谷歌学者 ]
阿诺德·K。; 博多利。; 科普J。; Schwede T。 生物信息学 2006, 22, 195. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Sharkey N.A。; Leach K.L。; 布隆伯格下午。 程序。 国家。 阿卡德。 科学。 美国。 1984, 81, 607. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Foloppe N。; Sagemark J。; Nordstrand K。; 伯恩特·K·D。; 尼尔森L。 分子生物学杂志。 2001年,第310页,第449页。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Vanommeslaeghe K。; 海彻E。; Acharya C。; 昆都S。; 钟S。; 垫片J。; Darian E。; Guvench O。; 洛佩斯·P。; 沃罗比约夫一世。; 麦克雷尔A.D。 J.计算。 化学。 2010, 31, 671. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
汉弗莱·W。; Dalke A。; 舒尔滕·K。 摩尔图形模型。 1996年,14,33。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Klauda J.B。; 布鲁克斯·B·R。; 牧师R.W。 化学杂志。 物理学。 2006, 125, 144710. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
关联数据 本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。