肿瘤经常表现出对胚胎发育和组织稳态至关重要的信号通路的不适当激活。 这些途径不仅有助于肿瘤细胞增殖和逃避细胞死亡的能力,而且还改变了细胞的可塑性。 胰腺导管腺癌(PDAC)符合这种模式,通常表现为胚胎信号通路的重新激活,如转化生长因子-β(TGFβ)、Notch、Hedgehog(Hh)和 Wnt–β-连环蛋白 信号(参见 参考1 PDAC的分子遗传学综述)。
与人类肿瘤不同,如髓母细胞瘤,异常的Hh信号足以导致疾病发展 2 , 三 或结肠癌 4 在这种情况下,Wnt–β-catenin的放松调控可以代表一个起始事件,遗传实验表明,Hh和Wnt–α-catenin信号的错误调控不足以驱动PDAC的发展。 相反,通过靶向胰腺癌基因表达驱动PDAC小鼠模型的分析 KRAS公司 提示Hh和Wnt–β-catenin活性的时间和空间控制都与指定可进展为PDAC的细胞系有关。 我们回顾了KRAS改变胰腺细胞命运的能力,以及Hh和Wnt–β-catenin信号的时间和位置如何促进PDAC的发展。
突变体KRAS推动PDAC开发 大量工作已用于确定PDAC的分子基础。 虽然一些(约2–10%)PDAC似乎与遗传因素有关 5 , 6 ,大多数与一组基因的高频体细胞突变有关,包括编码小GTPase蛋白KRAS的基因 7 和肿瘤抑制剂INK4A 8 ,p53( 参考9 , 10 )和 座椅模块组件4 ( 参考11 ). 在这些经常观察到的变化中,关键是要注意KRAS突变在人类PDAC中几乎是普遍存在的(>95%)。 PDAC中发现的KRAS突变导致蛋白质锁定在组成活性状态,无法水解GTP,从而促进下游效应器的持续信号传递(在 参考12 ). 尽管大规模的基因组研究正在扩大对PDAC中发现的更广泛突变的了解 13 研究上述“经典”基因在细胞培养和动物模型中的功能,对PDAC的维持和进展有了相当大的帮助。 PDAC与非侵袭性癌前病变有关,这些病变被认为是疾病的前兆 14 ( 方框1 ). 胰腺上皮内瘤变(PanINs)是最常见、研究最广泛的假定前体。 它们在组织学上分为细胞和核异型性增加的三个阶段 15 ( 图1 ). 分子研究表明,PanIN阶段与突变频率和种类的增加有关 16 例如,PanIN1病变经常具有突变的KRAS(估计为15-40% 17 )但很少有p53或SMAD4突变。 PanIN3病变更可能表达突变的KRAS、p53和SMAD4( 参考18 , 19 ).
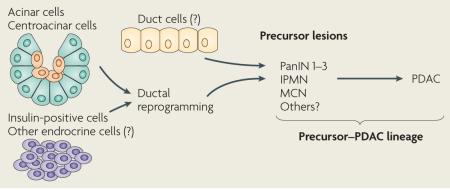
方框1。 KRAS在胰腺导管腺癌逐步进展中的必要性和充分性。 黏液性囊性肿瘤(MCN)、导管内乳头状黏液性肿瘤(IPMN)和胰腺上皮内瘤变(PanIN) 1 被认为是胰腺导管腺癌(PDAC)的前驱期。 这种观点得到了KRAS驱动的小鼠模型的支持,在这些模型中,癌前病变先于PDAC的发展( 表1 ). 事实上,在开发PanIN(人类最常见的假定前体病变)和PDAC的模型中,PanIN病变的严重程度增加,并且随着小鼠年龄的增长,PDAC变得更加普遍。 因此,在这篇综述中,我们将这种细胞命运称为PanIN–PDAC谱系(在下图中表示为前体–PDAC谱系)。 然而,其他小鼠模型表明,IPMN和MCN可能是PDAC开发的并行路径。 所有三个病变都有导管标记物的表达,因此如果它们来自非导管室,如腺泡、中心腺泡细胞或内分泌细胞,则需要重新编程为导管谱系。
尽管突变型KRAS足以在小鼠中启动PanIN–PDAC谱系,但有一些证据表明可能没有必要。 尽管经常观察到KRAS突变,但在早期人类PanINs中并不普遍。 此外,在小鼠中,由于Hedgehog配体在胰腺发育过程中过度表达,类似早期PanINs的病变可能会发生 57 和慢性炎症 120 KRAS突变在晚期PanINs和PDAC中变得越来越频繁,这导致了一个重要但尚未解决的问题,即什么时候解除KRAS活性对疾病进展是必要的。
图1。 KRAS是胰腺导管腺癌发生和发展的主要调节因子。
构成活性KRAS(由 喀斯特 G12D系列 或 喀斯特 G12伏 突变)足以引发胰腺上皮内瘤变(PanIN)和胰腺导管腺癌(PDAC)的发展。 PanINs可分为细胞异型性增加的三个阶段,在人类中,已发现其具有越来越多的突变(方框中显示了常见突变)。 上皮的变化与基质的促结缔组织增生性变化相匹配。 在小鼠模型中,通过激活胚胎胰腺祖细胞中的突变KRAS,重现了人类PanIN谱,随后发展为PDAC。 消除通常在人类疾病中失活的肿瘤抑制因子可显著降低PDAC潜伏期(仅举了一组有限的例子)。 KRAS在某些成体细胞类型中被特异激活的小鼠模型表明,腺泡和胰岛素阳性细胞均可产生PanINs,在某些情况下,PDAC取决于组织损伤和肿瘤抑制因子失活。 对于这些细胞类型,需要重新编程为“导管”细胞类型才能假定PanIN–PDAC血统。 由于尚未专门评估着丝粒细胞和导管细胞在KRAS控制下重新编程为能够成为PDAC的谱系的能力,因此对它们显示出问号。然而,在实现特定靶向之前,不能排除它们是前体PDAC谱系的来源。 经过许可,从以下位置修改了图 参考128 ©(2000)美国癌症研究协会。
由于其在PDAC中的普遍频率 KRAS公司 被认为是该病的起始遗传损伤。 然而,由于转基因方法的局限性,最初审核突变KRAS启动PDAC进展的充分性的工作受到阻碍。 腺泡和导管启动子下突变KRAS的表达导致导管病变,使人想起PanIN和腺泡和导管混合癌 20 或导管周围炎症 21 分别是。 然而,这两种模型均未导致PDAC或前体病变逐步进展的忠实再现。 尽管尚不清楚为什么这些模型未能再现人类疾病的进展,但它们可能受到了超生理性KRAS输出或在不适当的细胞类型或发育阶段激活KRAS的阻碍。 直到Cre诱导的条件等位基因(lox-stop-lox)的发展,突变KRAS驱动PDAC的能力才被成功研究 喀斯特 G12D系列 ( LSL-Kras公司 G12D系列 ))针对内源性 喀斯特 轨迹 22 从而允许在时间和空间控制下表达构成性活性KRAS。 该工具消除了可能存在的混淆细胞对过度表达的反应的问题,如突变体的转录 喀斯特 等位基因取决于内源性 喀斯特 发起人。 最初,小鼠表达 LSL-Kras公司 G12D系列 将等位基因与在关键胰腺祖细胞基因启动子控制下表达Cre重组酶的小鼠杂交:胰腺和十二指肠同源盒1( Pdx1型 )和 第48页 (也称为 Ptf1a公司 )从而将突变KRAS靶向发育中胰腺的大多数细胞。 少量 Pdx1-芯; LSL-Kras公司 G12D系列 和 p48核心; LSL-Kras公司 G12D系列 老鼠( 表1 )在一年的时间里开发了PDAC 23 此外,早期PanIN普遍具有渗透性,随着年龄的增长,观察到类似于整个人类PanIN光谱的病变。
表1。
遗传模型 表型 参考
Pdx1-芯; LSL-Kras公司 G12D系列 p48Cre; LSL-Kras公司 G12D系列
通用渗透剂PanIN开发。 年龄依赖性增加 病变严重程度和偶发PDAC,潜伏期长
23
Pdx1-芯; LSL-Kras公司 G12D系列 ; Cdkn2a公司 flox/flox公司
加速PanIN和PDAC开发
31
Pdx1-芯; LSL-Kras公司 G12D系列 ; Trp53基因 172H兰特/+
加速了PanIN和转移性PDAC的发展。 PDAC是 常规局灶性,分化良好,基因组广泛不稳定
33
Pdx1-芯; LSL-Kras公司 G12D系列 ; Smad4公司 flox/flox公司 p48核心; LSL-Kras公司 G12D系列 ; Smad4公司 flox/flox公司
IPMN和PDAC的发展
34
p48核心; LSL-Kras公司 G12D系列 ; Tgfbr2型 flox/flox公司
加速PanIN和PDAC开发。 延迟进一步降低 与杂合性比较
35
Pdx1-芯; LSL-Kras公司 G12D系列 ; Trp53基因 flox/flox公司
加速PanIN和差异化PDAC开发
32
Pdx1-芯; LSL-Kras公司 G12D系列 ; 墨水4a flox/flox公司
加速开发PanIN和低分化PDAC
32
p48核心; LSL-Kras公司 G12D系列 ; Smad4公司 flox/flox公司
开发MCN和PDAC。 潜伏期缩短,杂合子缺失
36
Pdx1-芯; LSL-Kras公司 G12D系列 ; Smad4公司 flox/flox公司
加速PanIN、IPMN和PDAC开发
37
雀巢Cre; LSL-Kras公司 G12D系列
外分泌衍生PanIN的发展
129
p48 Cre基因; LSL-Kras公司 G12D系列 ; Ela-Tgfa公司 Ela-Core公司 应急响应小组 ; LSL-Kras公司 G12D系列 ; Ela-Tgfa公司
加速PanIN、IPMN和PDAC开发
116
Ela-Ta; tetO-Cre; LSL-Kras公司 G12伏
腺泡和着丝粒衍生PanIN和PDAC的开发 什么时候 喀斯特 G12伏 在发育过程中或出生后激活。 慢性 PanIN和PDAC发育所需的胰腺炎 喀斯特 G12伏 在成体细胞中激活
24
Ela-Core公司 ERT2系统 ; LSL-Kras公司 G12D系列 薄雾1-Cre ERT2系统 ; LSL-Kras公司 G12D系列
腺泡衍生蛋白的研制
25
Pdx1-芯 应急响应小组 ; LSL-Kras公司 G12D系列 ; R26缺口 网络接口卡 Ela-Core公司 应急响应小组 LSL-Kras公司 G12D系列 ; R26缺口 网络接口卡
加速腺泡衍生PanIN的开发
26
薄雾1-Cre ERT2系统 ; LSL-Kras公司 G12D系列 ; 米斯特1 −/−
加速腺泡衍生PanIN的开发
27
Pdx1-芯; LSL-Kras公司 G12D系列 ; Tif1γ flox/flox公司
IPMN的快速发展
38
Ela-Core公司 应急响应小组 ; CAG-lox-GFP-lox-Kras公司 G12伏
慢性胰腺炎的快速发展 癌与PDAC
28
最近,这些原始模型已经被修改,以确定当突变型KRAS表达时,哪些胰腺细胞类型可以发展为PDAC。 通过使用使用诱导Cre的策略,可以激活 喀斯特 G12D系列 在特定的成体细胞群体中,PDAC虽然表现出导管特征,但它可能不一定来自导管室 24 – 30 ( 图1 ; 表1 ). 因此,突变KRAS是PanIN–PDAC“谱系”的关键决定因素,能够驱动胰腺细胞从终末分化为导管样命运,最终导致PDAC。
此外,KRAS驱动的模型与最常见的失活肿瘤抑制因子的功能丧失等位基因相结合,包括 墨水4a , Trp53基因 和TGFβ信号级联的各种成分 31 – 38 (模型总结如下 表1 )揭示了这些途径限制了KRAS导向的PDAC开发。 有趣的是,消除不同的肿瘤抑制因子可以显著改变发生的前体病变类型和恶性疾病的最终分化状态。 值得注意的是,这些模型中的细胞同时受到KRAS激活和肿瘤抑制功能丧失的影响。 这可能与人类肿瘤的进展形成对比,在人类肿瘤的发展过程中,KRAS突变似乎发生在疾病发展的早期,细胞随后承受选择压力并累积进行性肿瘤抑制因子丢失。 观察到,强制肿瘤抑制因子丢失(可能发生在人类疾病的自发发展过程中)会改变PDAC的发展过程,这一观察结果表明,疾病依赖于信号通路的特定和顺序调节,其方式与正常发展类似。 人类肿瘤和小鼠模型的特征表明,这种进展不仅取决于损害肿瘤抑制因子,还取决于其他信号通路异常活动的发展,包括决定发育过程的信号通路。 本综述的其余部分集中于其中两种途径:Hh和Wnt–β-catenin信号传导。
PDAC中的Hh信号 Hh信号在胚胎发育中起着至关重要的作用,并与体内平衡有关(影响再生 39 , 40 和干细胞维护 41 – 43 )和成人器官的疾病发展(有关依赖于该途径的生物化学和发育生物学的综述,请参阅 参考44 , 45 ). Hh信号由三种通常是组织特异性的分泌配体家族介导(声波刺猬( SHH公司 ),印度刺猬( IHH公司 )和沙漠刺猬( DHH公司 )). Hh配体通过与12通路跨膜受体结合激活靶细胞中的信号传导( PTC公司 ). 在缺乏配体的情况下,PTC抑制7通路跨膜受体的活性,使其平滑( SMO公司 ). 配体结合使PTC失活,从而激活SMO。 SMO激活反过来刺激Hh信号的下游细胞内成分,从而导致Gli转录因子活性形式的细胞质积累和核定位。 在哺乳动物中发现了三个Gli家族成员, GlI1公司 , GlI2公司 和 甘氨酸 ,并且每个都具有独特的属性。 GlI1仅以活性形式存在,GlI2也主要作为激活剂发挥作用,因为其阻遏物形式不如其激活形式稳定 46 ,而GlI3主要作为阻遏物发挥作用,因为其阻遏形式比其瞬时全长形式更稳定 47 改变这种激活和抑制Gli蛋白的平衡可以导致诱导转录靶点,从而驱动Hh依赖性表型,例如通过表达cyclin D1进行增殖( 参考48 )和MYC 49 ,通过促进BCL2的表达来避免凋亡( 参考50 )通过表达Forkhead家族转录因子实现细胞分化 51 , 52 此外,Hh信号的大小和持续时间可能会因通路的正调控因子(如GlI1)和负调控因子(例如PTC)的变化而改变。
Hh信号在肿瘤发生中的作用与对Hh生物化学及其对正常发育的贡献的理解平行出现。 肿瘤中的去调节Hh信号可以大致分为两类,这取决于信号级联中通路激活的水平(由 参考53 ). 第一类是由关键调节蛋白中的细胞自主突变定义的; 例如,PTC的失活突变或SMO的激活突变,如在基底细胞癌中观察到的突变 54 , 55 和髓母细胞瘤 56 Hh驱动的第二类肿瘤,包括乳腺癌、结肠癌、前列腺癌和PDAC,其特征是配体表达不当。 在人类PDAC中观察到异常的Hh配体表达频率很高(约75%),并且在整个疾病进展过程中都可以检测到,从早期的PanINs开始 57 ,一种在突变KRAS驱动的小鼠模型中重述的表达模式 33 PDAC细胞中的.Hh信号最初被认为是以自分泌方式激活的。 然而,尽管靶向SMO水平的通路的化学抑制剂(如环胺)或Hh配体可以减少异种移植的原发性人类肿瘤和细胞系的肿瘤负担和转移,而SMO抑制影响突变KRAS突变小鼠模型的生存和肿瘤发展 58 – 61 最近的证据表明,上皮性PDAC细胞对Hh配体没有反应,并且对配体抑制不起作用 62 , 63 相反,PDAC中的Hh信号似乎涉及肿瘤微环境中的一个配体依赖性成分,在该微环境中经典级联被激活,而肿瘤上皮中的配体依赖模块中Gli活性被解除调节( 图2 ).
图2。 胰腺导管腺癌中的刺猬信号。
尽管胰腺导管腺癌(PDAC)上皮过度表达Hedgehog(Hh),但基质细胞(包括癌相关成纤维细胞、浸润性骨髓源细胞和内皮细胞亚群)中的配体依赖性典型信号通过补片(PTC)-平滑(SMO)轴被激活。 反过来,这些细胞直接增殖或产生可能促进肿瘤细胞生长的因子(可能通过分泌生长因子或通过改变细胞外基质(ECM)组成)和旁分泌方式的血管生成(已确定的因子已指出)。 此外,癌相关成纤维细胞和其他Hh反应细胞可能产生细胞因子和其他分子,与浸润免疫细胞进行沟通。 相反,通过这一典型途径的自分泌激活似乎并不发生在肿瘤上皮中。 Gli活性部分是通过其他信号途径激活GLI1来维持的,例如突变KRAS表达和转化生长因子-β(TGFβ)信号。 血管生成素1; 胰岛素样生长因子; 基质金属蛋白酶9; VEGFA,血管内皮生长因子A。
强健的促结缔组织增生反应是PDAC的一个特征。 PDAC的促结缔组织增生基质由一系列复杂的细胞类型组成,包括癌相关成纤维细胞(CAF)、炎性细胞和肿瘤相关血管。 越来越多的证据表明,旁分泌Hh信号在支持肿瘤上皮和基质之间的促肿瘤原性通讯中起着关键作用,尤其是在CAF方面。 CAF被广泛认为是肿瘤发生的促进剂 64 重组实验中,来自不同癌症类型的肿瘤细胞与CAF混合并作为异种移植物生长,结果表明CAF可以促进永生化上皮的转化(例如SV40-永生化前列腺细胞 65 )促进肿瘤细胞生长 66 PDAC衍生的CAF对异种移植PDAC细胞的生长具有类似的促进作用 67 – 69 为了支持Hh配体在促进肿瘤生长中的旁分泌作用,Yauch及其同事 62 结果表明,招募到人类PDAC细胞系异种移植的基质细胞具有活跃的Gli信号,用SMO抑制剂处理的异种移植显示小鼠Hh靶基因的表达降低,但不降低移植衍生人类基因的表达。 Yauch及其同事使用小鼠胚胎成纤维细胞(MEFs)作为CAFs在重建实验中的替代物 62 结果表明,与表达SMO的细胞相比,缺乏SMO的MEF在促进肿瘤生长方面的效率显著降低。 此外,贝利和他的同事们 70 结果表明,将人胰腺成纤维细胞与转化的SHH过度表达的人导管细胞联合移植,可增加异种移植物的生长。 尽管旁分泌Hh信号明显有助于PDAC的维持,但旁分泌功能、CAF衍生因子的确切性质仍不清楚。 然而,分析实验表明,它们可能包括一些已知的促肿瘤因子,如胰岛素样生长因子 62 .Hh配体信号不仅可以驱动CAF产生致癌因子,还可以支持激活的CAF表型,其特征是平滑肌肌动蛋白(肌成纤维细胞状态的标志物)的表达以及细胞外基质蛋白(如纤维连接蛋白和I型胶原)的产生 71 后者已被证明能增强PDAC细胞的增殖和侵袭力 72 – 75 用SHH治疗人胰腺成纤维细胞可增加平滑肌肌动蛋白的表达,异种移植细胞中SHH配体的过度表达可增强招募的宿主成纤维细胞中I型胶原和纤维连接蛋白的表达 71 此外,Hh配体促进CAF增殖并刺激CAF迁移 71 因此,上皮衍生的Hh配体可启动支持CAF的前馈回路,并刺激作用于肿瘤细胞的因子的产生( 图2 ). 随着PanINs严重程度的加重,Hh配体表达增加 57 这可能是在PDAC进展期间扩展和维护CAF的机制。
最近的报告表明,旁分泌Hh信号的作用可能不仅限于肿瘤细胞–CAF轴,而且旁分泌Hh-配体信号也可能影响肿瘤相关血管系统。 bailey及其同事 70 表明SHH的表达增加了转化人胰腺导管细胞异种移植的血管生成。 这种效应部分依赖于配体信号传导,因为用阻断抗体对Hh配体进行功能性抑制可阻断表达Hh配子的异种移植瘤细胞的淋巴管生成 70 事实上,这种效应可能是直接的,因为Hh配体能够激活淋巴管内皮细胞的迁移和扩张 70 也可能通过间接机制发生,如诱导促血管生成因子; 例如,胰岛素样生长因子1(IGF1)和血管生成素1( 参考76 )由招募到肿瘤微环境中的骨髓源细胞、血管内皮生长因子A(VEGFA)和基质金属蛋白酶9(MMP9)产生 70 由CAF制作。
SMO抑制对KRAS驱动小鼠模型疾病进展的影响进一步反映了旁分泌Hh配体信号在PDAC中的重要性。 奥利夫和同事 61 最近证明 Pdx1-芯; LSL-Kras公司 G12D系列 ; Trp53基因 172H兰特/+ 或 Pdx1-芯; LSL-Kras公司 G12D系列 ; Trp53基因 R270小时/+ SMO拮抗剂和 吉西他滨 ,一线PDAC化疗药物 77 , 78 ,不仅提高了生存率和减少了转移,而且显著降低了肿瘤的成纤维细胞成分。 有趣的是,SMO抑制剂治疗可暂时增加肿瘤血管密度,并增加到达肿瘤细胞的吉西他滨浓度。 尽管这是一个令人惊讶的发现,因为数据表明基质Hh信号具有促血管生成作用,但它可能反映出PDAC中观察到的明显促肿瘤新生血管减少依赖于基质和上皮细胞之间的平衡。 尽管如此,这些研究表明基质Hh信号可能是一个有用的治疗靶点,可以与其他药物并行研究。
如果PDAC上皮中缺乏配体介导的典型信号,那么Hh通路是否在肿瘤细胞的进化中起直接作用? 可能是这样的,因为一些研究表明,活性Gli蛋白支持上皮PDAC细胞的增殖和活性 79 – 81 目前,支持Gli信号的非规范激活涉及两个信号级联。 通过小干扰RNA(siRNA)直接靶向突变KRAS或通过化学抑制剂靶向其下游RAF–MAPK效应通路降低Gli靶基因的转录和肿瘤细胞的生长 81 , 82 此外,阻断PDAC细胞中TGFβ信号传导抑制Gli活性和细胞生长 83 TGFβ对Gli信号的调节提出了关于三种不同形式Gli蛋白的相对浓度如何促进Gli输出的问题。 用TGFβ处理来自KRAS驱动的小鼠PDAC的细胞株可增加GlI1和GlI3的表达,并且GlI3被诱导到比GlI1更高的水平( 参考81 ). 这令人惊讶,因为GlI3主要作为Gli靶点的阻遏物。 然而,测序分析表明 GLI3公司 和相关基因 GLI4型 在PDAC中发生高频突变 13 因此,上皮细胞Gli信号的调控可能不仅通过非规范上游信号发生,还可能涉及Gli蛋白的新相互作用和功能。
Hh信号和PDAC开发 由于Hh信号传导在PDAC的进展和维持中似乎很重要,一个关键问题是它如何促进疾病的发生。 已经使用了几种遗传方法来确定单独调节Hh信号以及在突变KRAS的情况下如何改变PDAC开始的过程(这些模型总结于 表2 ). Hh信号和PDAC启动之间的关系首次在实验中被发现,这些实验旨在确定配体介导的Hh信号如何指导胃肠道发育。 一些发育研究表明,正常的肠道发育依赖于Hh配体的严格控制表达。 例如,在胰腺发育早期表达SHH的转基因小鼠 Pdx1页 发起人( Pdx1小时 ) 84 显示接近完全的胰腺发育不全,并取代正常胰腺,具有嵌入扩张的间充质室中的导管结构,表达肿瘤相关基质的分子标记 84 有趣的是,导管残余物在形态学上与早期人类PanIN1–2损伤相似,其中一些转基因小鼠也发生了自发的胰腺特异性KRAS突变,这表明不适当的配体表达可能会促进PDAC启动过程中组织结构和信号的改变 57 然而,这些动物在出生后不久(3周)死亡,这妨碍了对Hh配体信号单独驱动PDAC能力的评估。 为了避免异常配体信号传导的发育后果,开发了旨在测试Hh信号级联的细胞内模块是否可以驱动PDAC的模型。 首先,携带 CLEG2电缆 转基因允许Cre诱导的条件表达一种缺乏氨基末端阻遏物结构域的主要活性GlI2蛋白,该转基因与表达 Pdx1-芯 司机 85 约三分之一 Pdx1-芯; CLEG2电缆 小鼠发生胰腺肿瘤 85 由未分化的纺锤形细胞组成,在分子或组织学上与PDAC不相似。 因此,尽管Gli信号在胰腺中可能致癌,但仅此一项在推动PDAC发展方面是无效的。
表2。 小鼠胰腺Hedgehog和β-catenin失调模型
遗传模型 表型 参考
刺猬
Pdx1小时
萎缩胰腺显示类似于PanIN1和2嵌入的病变 肠样基质
57
Pdx1-芯; CLEG2电缆
大的未分化胰腺肿瘤的发展
85
Pdx1-芯; CLEG2; LSL-Kras公司 G12D系列
加速PanIN发育和未分化“CLEG2样”肿瘤
85
CAGGS-核心; R26-烟雾2
MCN样病变的发展
130
p48核心; LSL-Kras公司 G12D系列 ; Trp53基因 弗洛克斯/+ ; 平滑(Smo) flox/flox公司
与相比,PanIN和PDAC开发没有差异 p48核心; LSL-Kras公司 G12D系列 ; Trp53基因 弗洛克斯/+ 老鼠
81
p48核心; LSL-Kras公司 G12D系列 ; 烟雾2
与相比,PanIN开发没有加速 p48核心; LSL-Kras公司 G12D系列 老鼠
63
β- 连环蛋白
Pdx1-芯 迟到 ; Ctnb1号机组 外显子3/+
腺泡增殖和产后胰腺肿大。 无肿瘤发生
102
Pdx1-芯; 亚太区 flox/flox公司
腺泡增殖和产后胰腺肿大。 无肿瘤发展
103
p48 Cre基因; Ctnb1号机组 外显子3/+
类人实性假乳头状肿瘤的发展
104
p48核心; Ctnb1号机组 外显子3/+ ; LSL-Kras公司 G12D系列
类似于人导管内管状肿瘤的肿瘤的发展
104
鉴于PDAC中激活KRAS突变的频率很高,并且在完全转化的PDAC细胞系中发现了KRAS的突变和持续的Gli活性,KRAS和Gli信号协同驱动PDAC启动和发育的能力通过相互交叉来确定 Pdx1-芯; CLEG2电缆 带有 LSL-Kras公司 G12D系列 动物。 在出生后3到6周内,这些动物迅速发展为PanIN病变,包括具有晚期2级和3级病变特征的病变 85 该模型中的PanIN病变也表现出异常的Hh配体表达,并伴有扩大的增生性间质室,进一步将Hh配子信号与间质激活联系起来。 然而 Pdx1-芯; CLEG2; LSL-Kras公司 G12D系列 小鼠没有发展成PDAC,而是发展成一个来源不明的未分化肿瘤,类似于在 Pdx1-芯; CLEG2电缆 老鼠。
PanIN位于 Pdx1-芯; CLEG2; LSL-Kras公司 G12D系列 该模型始终显示Hh配体的异常表达,进一步表明配体依赖信号在PDAC发育中的作用。 为了确定这种作用是否依赖于上皮细胞中的配体依赖信号,Steveaux及其同事生成了 p48 Cre基因; LSL-Kras公司 G12D系列 ; 平滑(Smo) flox/flox公司 老鼠 81 从而使胰腺祖细胞对Hh配体不敏感。 令人惊讶的是,这些小鼠的PanIN和PDAC发育速度与野生型SMO对照组相当。 同样,这两种基因型都出现持续的上皮配体过度表达和同等水平的Hh靶基因激活。 此外,在来源于 平滑(Smo) -不足和 平滑(Smo) -野生型肿瘤导致细胞凋亡增加和细胞生长下降。 因此,该模型表明,异常配体表达和上皮细胞Gli信号传导有助于PDAC的启动和进展,但它们是不耦合的,并且是独立进化的。
综上所述,这些小鼠模型表明,非典型上皮细胞Gli信号和基质配体依赖信号与KRAS协同作用,驱动PanIN启动和进展为PDAC。 然而,重要的问题仍然存在。 Hh配体明显参与指导肿瘤微环境,尽管哪些基质Hh靶基因是维持CAF和肿瘤基质其他成分的关键,以及哪些因素对上皮产生影响,包括影响分化,目前尚不清楚。 此外,上皮Gli活性似乎参与了PDAC的进展,并由于经典Hh配体-SMO依赖性途径以外的途径的活性而发展。 更好地了解这些非经典途径(包括KRAS和TGFβ信号传导)如何激活Gli信号传导,以及哪些上皮Gli靶点对维持肿瘤生物学至关重要,可能会提供额外的治疗靶点,与基质细胞中Hh配体刺激激活的因子平行。 另一种治疗可能性是确定可能与Hh信号并行作用的其他发育信号途径。 Wnt–β-catenin信号通路是一种已被证明对PDAC启动和进展具有实质性影响的候选途径。
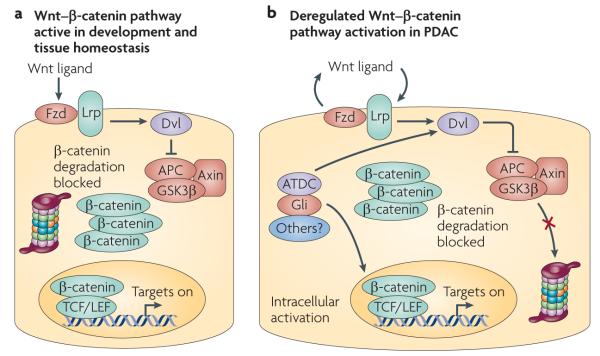
PDAC中Wnt–β-catenin信号 与Hh信号传导一样,Wnt–β-catenin信号传导是一种重要的胚胎信号传导途径,对几个器官的增殖、形态发生和分化是必需的(有关综述,请参阅 参考86 ). β-catenin信号转导在别处已得到很好的评价 87 (总结于 图3a ). 简单地说,Wnt配体与Frizzled蛋白家族的受体和共受体结合。 配体结合导致细胞质蛋白复合体(包括腺瘤性息肉病大肠杆菌(APC)和axin)失活,促进β-catenin的蛋白酶体降解,导致其细胞质积累和核定位。 在细胞核中,β-catenin与TCF/LEF因子结合以激活靶基因。 已在哺乳动物中鉴定出19种Wnt配体,已知它们在与Frizzled–LRP联合受体复合体结合时激活典型(β-catenin-dependent)和非典型(β-catenin-independent)信号转导级联。 在本节中,我们将重点介绍典型途径,因为该途径与PDAC的发展有关。 然而,非规范Wnts如WNT5a在PDAC中被解除管制,可能在PDAC生物学中起作用 88 , 89 .
图3。 胰腺导管腺癌中典型的Wnt–β-catenin信号。
一 |在正常发育过程中,典型的Wnt–β-catenin信号依赖于激活受体(Frizzled(Fzd)–Lrp复合物)的分泌配体(Wnt配体),该受体阻断由破坏复合物(包括腺瘤性息肉病大肠杆菌(APC)、axin、糖原合成酶激酶3β(GSK3β))促进的β-catening蛋白体降解 和其他蛋白质)。 b条 |β-catenin在胰腺导管腺癌(PDAC)中积聚较为常见。 累积的β-catenin可以与TCF/LEF共因子一起转位到细胞核并激活靶基因。 目前,PDAC中持续β-catenin积累和活性的主要机制尚不清楚。 有证据表明自分泌(由于上皮衍生Wnt配体)和细胞自主激活(通过Gli信号和基因,如共济失调毛细血管扩张症D组相关基因( ATDC公司 ),激活Dvl)。 基质细胞和细胞外基质也可能促进β-连环蛋白的积累。
编码典型信号级联调节蛋白的基因突变与许多肿瘤类型相关,在某些情况下,如结肠癌,可能会引发遗传损伤。 负调控因子如APC的功能丧失突变 4 β-catenin自身的获得性功能突变 90 以与PTC和SMO突变类似的方式激活组成性、配体依赖性β-catenin信号。 像这些Hh途径突变一样,经典β-连环蛋白调节模块的遗传损伤在PDAC中很少见 91 – 93 因此,β-catenin在PDAC中的作用一直存在争议。
然而,最近的研究已经开始巩固Wnt–β-catenin信号在PDAC中的重要作用。 虽然β-catenin的积累不是该病的普遍特征,但在PanIN病变和PDAC中观察到β-catentin的核(10–60%)和细胞质积累(25–65%) 94 – 96 功能性证据也在积累,表明β-catenin在PDAC维持和进展中起支持作用。 用siRNA抑制β-catenin可显著抑制PDAC增殖并增加凋亡 96 此外,细胞质和细胞核β-连环蛋白水平的升高与PanIN分级和侵袭性PDAC的发展相关 94 , 97 最近,共济失调-毛细血管扩张症组D相关蛋白(ATDC,也称为TRIM29)被证明有助于PDAC中β-连环蛋白的进行性积累和β-连环素靶基因的持续激活 97 抑制PDAC细胞系中的ATDC功能可降低β-catenin活性、肿瘤生长和转移。 因此,诸如 ATDC公司 可能包含一组非经典的参与者,在其他β-连环蛋白驱动肿瘤中正常观察到的突变缺失的情况下,影响PDAC中致癌的β-连环素活性。 有趣的是,ATDC介导β-catenin的积累 97 导致MYC的表达增加,MYC是其他组织中β-连环蛋白驱动转化的关键节点。 还有新的证据表明,β-catenin的积累和信号传递可以通过PDAC微环境中发生的旁分泌信号传递而增加。 最近的研究表明,PDAC细胞与生长在三维细胞外基质塞中的CAF混合后,胞质β-catenin积累增加 98 I型胶原的生长促进核β-连环蛋白的积累和已知β-连环素靶基因的激活 75 .
除上述具体例子外,对PDAC中β-catenin积累的一般机制了解甚少(总结于 图3b ). 例如,尽管β-连环蛋白的积累与疾病的严重程度相关,但尚不清楚其积累是否取决于对肿瘤细胞过表达的Wnt配体的自分泌反应。 为了支持这一点,典型Wnt配体(包括WNT3和WNT8b)在PDAC样本中强烈表达 96 分泌型卷曲受体蛋白(SFRP,Wnt配体抑制剂)的强制表达降低了人类PDAC细胞亚群中β-catenin的转录活性 99 然而,另一种分泌型Wnt配体抑制剂DKK1的过度表达与更高级的PanINs和PDAC相关,并增加PDAC细胞的生长和运动 100 此外,在众多的β-catenin靶基因中,哪一个对PDAC的维持和进展至关重要目前尚不清楚。 因此,尽管经典的β-catenin致癌靶点在PDAC中可能很重要,但还需要更多的工作来确定是否以及在何种水平上β-catentin信号应该被靶向以获得潜在的治疗效果。
β-catenin不足以启动PDAC 越来越多的证据表明,β-catenin信号传导有助于PDAC的维持。 相比之下,β-catenin功能在PDAC启动过程中的作用最近才被分析。 三项研究调查了不受控制的β-catenin信号传导转化胰腺细胞的能力。 我们的研究使用了一个β-catenin等位基因,其中第三个外显子(编码对蛋白质降解重要的磷酸化位点)是漂浮的,可以通过Cre介导的重组消除 101 ,导致强制的、有条件的β-catenin稳定和信号传递。 当交叉到在所有胰腺祖细胞中表达Cre的Cre驱动线时- Pdx1-芯 早期 -稳定的β-catenin损害胰腺发育,导致严重外分泌发育不全和大囊肿的形成 102 有趣的是,这种对胰腺发育的影响与阶段和细胞类型有关。 将稳定的β-catenin等位基因与另一个Cre驱动系交叉, Pdx1-芯 迟到 Cre的表达被延迟并局限于腺泡和内分泌细胞,导致胰腺正常发育。 令人惊讶的是,在老龄小鼠中观察到外分泌胰腺在出生后扩张,这与腺泡增殖增加和β-连环蛋白积累有关。 有趣的是,尽管β-catenin在老年小鼠中持续积累,但未观察到肿瘤。 斯特罗姆和同事 103 生成 Pdx1-芯 表达关键破坏复合物成分APC的漂浮等位基因的小鼠( Pdx1-芯; 亚太区 flox/flox公司 )从而增强发育中胰腺细胞的典型Wnt信号。 这些小鼠出生后胰腺重量增加,这与外分泌增殖增强、核β-连环蛋白的年龄依赖性积累以及β-连环素靶基因的表达增加有关; 然而,这些小鼠在1岁时也没有发生肿瘤。 因此,这项工作支持了β-连环蛋白在外分泌胰腺中有效激活增殖的能力,并再次证明了β-连蛋白的转化能力在胰腺细胞中受到严格调控。
其他研究表明,当β-catenin信号在适当的发育阶段被激活时,它可以诱导胰腺转化。 使用a激活稳定的β-catenin p48芯 驾驶员( p48核心; Ctnb1号机组 外显子3/+ )不仅导致腺泡增殖和一些β-catenin靶基因的激活,还导致类似于人类实体假乳头状瘤(SPTs)的大型良性肿瘤的发生 104 这些SPT样肿瘤在人类中被证明携带β-连环蛋白激活突变 105 在形态和分子上与PDAC不同。 总之,这些模型表明,β-连环蛋白信号传导是外分泌增殖的关键介质,尽管它可以以时间和细胞类型依赖的方式诱导胰腺肿瘤发生,但似乎不足以启动PDAC。
鉴于KRAS在启动PDAC中的主要作用,一个重要的问题是KRAS是否能与β-连环蛋白信号协同作用,驱动PDAC的发展,正如在结肠癌等其他肿瘤类型中观察到的那样 106 – 108 令人惊讶的是, p48核心; Ctnb1号机组 外显子3/+ ; LSL-Kras公司 G12D系列 小鼠没有发生SPT样肿瘤,也没有发生PanINs或PDAC。 相反,他们发展出一种独特的导管肿瘤,类似于在人类中观察到的罕见导管内管状肿瘤(ITT) 104 因此,尽管PDAC表现出伴随的KRAS活性和β-catenin积累,但在发育中胰腺中这两条通路的同时激活似乎与PanIN–PDAC导管谱系的规范不相容。
β-catenin和KRAS在PDAC启动过程中意外缺乏协同作用,这表明发育信号通路必须在转化过程中的关键时间点调整到适当水平,以明确PanIN–PDAC谱系。 最近,发现胰腺炎是人类PDAC的一个潜在危险因素 109 – 111 在外分泌室中表达突变KRAS的小鼠中,加速PanIN和PDAC的发育,从而深入了解了β-catenin在这一过程中的作用。 这一发现也引发了一些有趣的发现,即胰腺中的哪些细胞具有重新编程为PanIN–PDAC病变的能力。
Acinar可塑性和PanIN–PDAC血统 与其他胃肠器官(如肠和结肠)不同,胰腺实质的转换速度很慢(例如,溴脱氧尿苷罕见地掺入健康成年小鼠胰腺细胞的DNA中就表明了这一点 112 ). 然而,胰腺具有强大的再生能力。 不同类型胰腺损伤(如化学性胰腺炎)后胰腺再生的研究 113 和部分胰腺切除术 114 )已经获得了关于重新填充受损胰腺室的细胞来源以及成人胰腺谱系固有可塑性的广泛知识。
在人类中,慢性胰腺炎是PDAC的潜在危险因素 109 – 111 最近,一些研究使用不同版本的KRAS驱动的小鼠PDAC模型来评估胰腺炎是否与PDAC的发生和发展有关。 这些研究不仅表明胰腺炎加速了PanIN和PDAC的发展,表明组织损伤和炎症与KRAS信号协同驱动疾病,还表明KRAS显著改变胰腺再生和可塑性。 第一项直接研究胰腺炎对PDAC发展影响的研究使用了一种小鼠模型,该模型允许多西环素诱导的组织学活性突变KRAS的暂时表达( 喀斯特 G12伏 )仅位于腺泡和中央腺泡室 24 类模型,其中KRAS由Cre在控制下诱导 Pdx1页 或 第48页 这些小鼠在胚胎发生期间或出生后不久KRAS的激活导致了频繁的PanIN病变和一些PDAC的发展。 虽然已经注意到人类和小鼠PanINs中存在腺泡标记物 115 该结果为PanIN–PDAC事件序列的非导管源提供了直接证据。 有趣的是,当突变型KRAS在出生后60天被激活时,这些小鼠对PanIN–PDAC的发育难以耐受。 因此,作者研究了慢性胰腺炎形式的持续性损害是否可以为PanIN–PDAC的发展提供一个宽松的环境。 他们通过长期使用蓝绿色素(一种胆囊收缩素类似物,可刺激腺泡细胞消化酶的早熟激活,导致胰腺自身消化和与炎症相关的细胞损伤)治疗诱发胰腺炎。 在对慢性胰腺炎的反应中,KRAS在成年期被激活的小鼠产生了高频率的PanINs和PDAC。 因此,本研究在功能上将胰腺炎和组织损伤与PDAC的发生和发展联系起来。
自本研究以来,其他研究小组已经验证了其关键发现,即KRAS可以将腺泡细胞重新编程为导管PanIN–PDAC谱系,并且胰腺炎可以有效地加速KRAS驱动的PanIN–PDAC的启动和进展。 使用其他可诱导的Cre细胞系,成人腺泡细胞已被证明对KRAS自发的导管重编程为PanINs敏感 G12D系列 在没有胰腺炎的情况下 25 , 26 尽管机制尚不清楚,但这些结果表明,不同的KRAS突变(在本例中为KRAS G12D系列 与KRAS相比 G12伏 )两者都使蛋白质具有组成活性,可能对外分泌细胞产生不同的生物和生化效应,这可能是由于Ras信号水平的差异所致。 事实上,有新的证据表明,腺泡到导管的重新编程进入PanIN–PDAC谱系依赖于突破关键的KRAS活性阈值( 图4 ). Ji和同事 28 显示来自表达KRAS的非转化胰腺细胞的活性GTP-结合KRAS水平增加 G12D系列 和源自KRAS的细胞 G12D系列 -驱动PDAC。 增强腺泡中KRAS活性的水平与PDAC中观察到的水平一致,导致幼年小鼠腺泡到导管的化生,这让人想起慢性胰腺炎。 随后,随着动物年龄的增长,PanINs和PDAC的发展 28 Siveke及其同事支持需要一个关键的KRAS活性阈值来启动导管重新编程 116 研究表明,TGFα的过度表达可以激活表皮生长因子受体下游的KRAS,与突变的KRAS结合,可以显著加速正常腺泡的消除以及PanINs和导管内乳头状黏液瘤(IPMN)的发展。 目前,哪些KRAS效应器对将腺泡重新编程为PanIN–PDAC谱系很重要尚不清楚; 然而,MAPK活动 29 和Akt信号 117 (Ras-activated PI3K下游)与腺泡到导管的可塑性有关。
图4。 发育信号通路和KRAS活性的关键时间阈值允许胰腺上皮瘤形成-胰腺导管腺癌的发生和发展。
高于关键阈值的KRAS活性可使分化的胰腺细胞(例如腺泡细胞)进入去分化的导管状态,这种状态持续存在于胰腺上皮瘤(PanIN)和胰腺导管腺癌(PDAC)中。 为了使这些去分化的导管细胞成为PanINs,β-catenin信号必须维持在关键的低水平以下。 然而,一旦PanIN状态建立,β-catenin信号被重新激活,同时Hedgehog(Hh)配体的表达增加,从而激活基质细胞中发生促结缔组织增生反应的靶基因。 发育中的肿瘤上皮细胞中的Gli活性独立于自分泌刺激产生。 虽然Gli活性可能在PanIN中是活跃的,但其在从PanIN向PDAC的进展中的作用尚不清楚。 最后,Gli活动可能对PDAC维护很重要。 经过许可,从以下位置修改了图 参考128 ©(2000)美国癌症研究协会。
其他研究证实了胰腺炎能够为PDAC前体提供一个宽松的环境,表明急性胰腺炎可以有效地促进PanIN的发育,在某些情况下,PDAC可以在外分泌室中表达突变KRAS的小鼠中进行 29 , 118 这些研究以及Guerra及其同事的研究 24 提示组织损伤是PDAC前体发育的宽松环境,改变胰腺再生可能与KRAS活性在启动导管重编程进入PanIN–PDAC谱系中发挥平行作用。 在野生型小鼠中,腺泡细胞在严重的氯化物诱导的胰腺炎后迅速再生 112 , 113 , 119 然而,腺泡细胞在损伤后立即发生实质性的形态和分子变化。 在这个再生阶段,腺泡细胞暂时重新激活胚胎胰腺发育的元素 113 ,常呈导管状形态 112 表达细胞角蛋白19,导管分化的标志物 29 然而,值得注意的是,在野生型动物中,这种去分化、导管状态不会固定,细胞会恢复腺泡分化。 这种有限的腺泡到腺泡的可塑性是为了应对其他刺激腺泡再生的损伤,如部分胰腺切除术 114 因此,在缺乏某些信号通路的异常激活的情况下,固定的腺泡至导管重编程(或腺泡至导管化生(ADM))是一种严格限制的分化命运。 它可以发生在野生型腺泡中 体内 ,但似乎需要持续的伤害。 斯特罗贝尔及其同事 120 注意到,患有7到10周慢性青霉素诱导的胰腺炎的小鼠出现了粘液化生病变(MML),具有早期PanINs(和新定义的胰腺导管腺体)的一些特征 121 ). 尽管大多数MML来源于导管或着丝粒细胞,但只有一小部分(约5%)是ADM.Desai及其同事的结果 114 还发现,小鼠胰腺导管结扎后出现ADM,这一过程的特点是腺泡严重凋亡,腺泡丢失时间延长。 然而,ADM可以通过其他信号通路的激活容易地被诱导,例如表皮生长因子(EGF) 122 和陷波信号 123 , 124 通过将腺泡暴露于基质金属蛋白酶7(MMP7) 124 以及最近发现的突变型KRAS的激活 25 此外,在发育过程中,外分泌细胞中PDX1的强制表达可诱导ADM 125 因此,当不适当激活时,一些与腺泡再生相关的因素(如Notch信号和PDX1表达)可以驱动腺泡到导管的重新编程,这表明必须严格调控发育信号通路以控制腺泡的可塑性。
综上所述,这些数据提出了一种可能性,即发育信号通路必须经过专门的调节,才能使KRAS将腺泡重新编程为导管PanINs。 我们最近的工作比较了正常腺泡和腺泡表达突变型KRAS对急性胰腺炎的再生反应,发现这是β-连环蛋白信号的情况 29 尽管表达腺泡的突变型KRAS对急性胰腺炎的反应类似于野生型腺泡,但其再生腺泡状态的能力被阻断。 相反,腺泡表达突变型KRAS持续表达导管标记物并重新激活胚胎发育元素,并迅速引起PanIN损伤。 由于胰腺发育的持续活跃、再生相关元素是PanINs的特征,这些数据表明假设去分化状态可能是KRAS驱动的PDAC发育中的一个速率限制步骤。 其他将突变KRAS与维持腺泡分化的基因突变结合在一起的小鼠模型也支持这种潜在的去分化作用。 例如,抑制肌肉、肠和胃1(MIST1)功能,这是一种在腺泡细胞中表达的转录因子,导致ADM表达急性胰腺炎后和KRAS驱动的导管重编程和PanIN形成过程中发现的去分化腺泡标志物 126 的确,结合 薄雾1 -KRAS突变敲除小鼠显著加速腺泡衍生PanINs的发育 27 支持去分化作用,作为KRAS驱动PDAC启动的一部分。
此外,β-catenin信号似乎是暂时性去分化、再生腺泡和持续性去分化导管重编程和PanIN形成腺泡之间的关键区别 29 在野生型小鼠中,β-catenin信号在青霉素诱导的胰腺炎后腺泡再生期间被激活。 然而,在表达突变型KRAS的小鼠中,β-catenin信号在同等时间点被阻断,这表示导管重新编程的早期阶段。 挑战同时表达突变KRAS和稳定β-连环蛋白的成年腺泡细胞,用盲囊素抑制PanIN的形成,反而导致导管结构异常,经常显示β-连环蛋白的核积聚 26 综上所述,突变体KRAS利用去分化腺泡状态驱动通常受限的导管PanIN谱系的能力似乎对发育信号通路的分子活性敏感,特别涉及低阈值的β-catenin信号( 图4 ).
因此,β-catenin在PanIN启动和进展为PDAC期间似乎具有相反的作用。 这些研究表明,经典β-catenin途径调节剂的突变在PDAC中主要不存在,因为它们可能会阻止KRAS将细胞启动为能够驱动PDAC的祖细胞样谱系的能力。 然而,如上所述,β-catenin在PanIN病变中积累并发出信号,支持PDAC的维持。 这些数据表明,β-catenin信号被调节,类似于正常器官规范期间所需的发育信号通路的时间调节,以有序的方式允许KRAS指定PanIN–PDAC谱系。 目前正在研究在腺泡重新编程进入PanIN谱系期间阻断β-catenin信号传导并在PanIN进展期间使其重新激活的确切机制。 最近的研究表明,由于其他发育信号通路的重新激活,可能会实现不同的β-catenin水平。 Siveke及其同事 119 研究表明,Notch受体激活抑制培养的腺泡细胞和急性胰腺炎后再生过程中的β-catenin活性。 此外,De la O及其同事 26 显示转基因NOTCH1的激活显著加速了ADM–PanIN。 这可能表明Notch信号(在急性胰腺炎反应中被激活)可能有助于在KRAS驱动的导管重编程期间维持允许的β-catenin信号阈值。 Notch在PanIN启动、PDA进展和β-catenin信号传导控制中的作用可能会被证明是复杂的。 例如,Hanlon及其同事最近证明了消除Notch1 Pdx1-芯; LSL-Kras公司 G12D系列 小鼠加速PanIN和PDAC的发展 127 然而,这些小鼠在Notch信号靶点激活方面既没有明显变化,也没有β-catenin积累增加。 因此,Notch和β-catenin在PanIN发育和PDAC进展中的相互作用可能取决于其他信号级联的输入。 例如,在PDAC上皮中非标准地激活Gli信号 96 和TGF-β信号已被证明支持PDAC细胞中的β-catenin信号 131 这些途径以及其他新的调节因子,如ATDC,可能有助于在建立PanIN–PDAC谱系后驱动PDAC中的β-catenin信号传导。
结论 KRAS驱动模型的出现为PDAC的启动和疾病进展提供了大量知识。 Hh和β-catenin信号明显影响PDAC的发育和维持。 下一个挑战是了解如何利用这些途径取得治疗成功。 这项工作的关键是通过已知的机制和发现新的调节器来确定每条通路是如何激活的分子基础。 此外,重要的是要了解这些途径如何相互作用,并比较和对比肿瘤上皮和微环境中的这些相互作用。 最后,必须努力确定哪些Hh和β-catenin靶点对维持增殖、活性和分化具有“关键任务”,并开发有效的方法(药理学和其他)来阻断这些关键信号节点。 随着这些重要目标的实现,它们为扭转PDAC的发育生物学远离恶性肿瘤打开了一扇机会之窗。
一目了然。
KRAS突变在人胰腺导管腺癌(PDAC)中几乎普遍存在。 突变型KRAS靶向胰腺的小鼠模型显示,KRAS信号足以将胰腺细胞重新编程为导管样谱系,能够通过癌前病变进展,并最终在令人想起人类疾病的阶段发展PDAC。
在KRAS驱动的PDAC小鼠模型中观察到的癌前病变的潜伏期、分化和类型对肿瘤抑制因子丢失敏感,这表明PDAC的进化依赖于信号通路的顺序调节。
PDAC的特点是频繁解除对胚胎信号通路的调控,包括Hedgehog(Hh)和Wnt–β-catenin信号通路。 最近的证据表明,在PDAC的发育和维持过程中,这些途径受到时间和空间的控制。
PDAC细胞经常表现出异常的Hh配体表达。 最近的研究表明,经典的配体依赖性信号在肿瘤微环境中的细胞中激活,以旁分泌方式支持肿瘤维持,但在肿瘤上皮中不支持。 然而,Gli转录因子水平的Hh信号在肿瘤上皮细胞中是活跃的,这是由该通路的非规范调节器所决定的。 在小鼠模型中,旁分泌配体活性和上皮胶质细胞信号传导似乎独立支持KRAS驱动的PDAC进化。
Wnt–β-catenin信号在PDAC中经常被激活,并有助于肿瘤细胞增殖和生物学。 允许Wnt–β-catenin解除调控的遗传模型表明,该途径可以转化胰腺细胞,但不足以驱动PDAC的启动。
小鼠模型显示,KRAS将细胞重新编程为可导致PDAC的导管样命运的能力对细胞分化和KRAS信号水平敏感。 胚胎信号通路的时间调控似乎在癌前重编程中发挥了作用,如KRAS驱动腺泡细胞去分化为PDAC前体病变期间Wnt–β-catenin信号的控制要求所示。
致谢 作者感谢R.Vanderlaan和A.Folias对手稿的批判性阅读和激发讨论。 M.H.胰腺癌实验室的工作得到了美国国立卫生研究院(NIH)(CA112537)和美国癌症研究协会胰腺癌网络(PanCAN)的资助。 S.C.W.由美国国立卫生研究院(NIH)资助,由国家癌症研究所(National Cancer Institute)和美国外科医生学院(American College of Surgeons Resident Research Scholarship)授予Ruth L.Kirschstein国家研究服务奖F32。