抗CTLA-4抗体诱导肿瘤病变内T reg细胞的选择性耗竭,其方式取决于肿瘤微环境中表达Fcγ受体的巨噬细胞的存在。
摘要
针对细胞毒性T淋巴细胞相关抗原4(CTLA-4)(T淋巴细胞表达的抑制性受体)的单克隆抗体治疗已成为转移性黑色素瘤的有效治疗方法。尽管存在争议,但目前的模型支持一种活性机制,即阻断CTLA-4对效应(T eff)和调节(T reg)T细胞的抑制活性,从而增强能够诱导肿瘤消退的抗肿瘤效应T细胞活性。然而,我们证明,T调节细胞室上的抗-CTLA-4抗体的活性是通过肿瘤病灶内T调节细胞的选择性缺失介导的。重要的是,T reg细胞的耗竭依赖于肿瘤微环境中表达Fcγ受体的巨噬细胞的存在,这表明T reg的细胞以依赖于环境的方式以反式形式耗竭。我们的研究结果揭示了对基于抗CTLA-4的癌症免疫疗法活性的进一步机制见解,并说明了局部肿瘤环境的特定特征对基于抗体的免疫调节疗法的最终结果的重要性。
全人类抗细胞毒性T淋巴细胞相关抗原4(CTLA-4)单克隆抗体Ipilimumab是第一种通过增强免疫抗肿瘤活性发挥作用的新型癌症治疗方法。两项关键的III期临床试验表明,伊普利单抗治疗的黑色素瘤患者的生存率显著提高(Hodi等人,2010年;Robert等人,2011年)这导致其最近获得FDA的批准。然而,尽管进行了深入调查,但行动机制仍不清楚。虽然最初的前提是抗-CTLA-4抗体(α-CTLA-4)通过阻断效应T细胞(T eff细胞;Krummel和Allison,1996年;Sutmuler等人,2001年),CD4的证明+Foxp3系列+调节性T细胞(T reg cell)表达高水平的CTLA-4,提示α-CTLA-4通过介导耗竭或影响其抑制活性直接影响T reg细胞室(Read等人,2000年,2006;Takahashi等人,2000年;Wing等人,2008年). 在这方面,我们最近证明,α–CTLA-4需要结合T eff和T reg细胞以诱导完全肿瘤保护(Peggs等人,2009年). 然而,一些出版物没有支持T调节细胞耗竭作为一种作用机制,相反,已经证明α-CTLA-4在次级淋巴器官中扩张T调节细胞(Quezada等人,2006年;施密特等人,2009年)还有血(Kavanagh等人,2008年)这进一步支持了CTLA-4限制T细胞增殖的观点。因此,α–CTLA-4直接影响T调节细胞室活性的机制尚不清楚。
α-CTLA-4介导的肿瘤排斥反应的一个共同特征是肿瘤内T eff与T reg细胞的比率增加(T eff/T reg细胞比率;Shrikant等人,1999年;Quezada等人,2006年;Kavanagh等人,2008年;Liakou等人,2008年;Chen等人,2009年;Curran和Allison,2009年;Waitz等人,2012年). 这种增加被认为是由于T eff优先于T reg细胞的扩张引起的,尽管尚不清楚为什么这种效应仅限于肿瘤微环境,以及为什么同时针对两个具有相反活性的细胞群体的抗体有利于效应T细胞功能并促进肿瘤排异。
在这里,我们通过关注控制肿瘤内T eff/T-reg细胞比率选择性增加的因素,进一步定义了α-CTLA-4抗肿瘤活性的潜在机制。通过追踪肿瘤特异性CD4+T细胞,我们发现α-CTLA-4增加了淋巴结中T eff和T reg细胞以及肿瘤中T eff细胞的绝对数量,同时选择性地减少了肿瘤中T reg细胞的绝对数量。T reg细胞的减少与肿瘤内存在表达FcγR的巨噬细胞相关的FcγRIV依赖性缺失以及肿瘤浸润T reg的细胞升高表面CTLA-4表达的机制一致。因此,α-CTLA-4阻断抑制信号,导致淋巴结中的T eff和T reg细胞以及肿瘤中的T eff细胞的扩张和积聚,但同时耗尽肿瘤浸润的T reg细胞,导致肿瘤内T eff/T reg细胞比率增加。总之,这些数据解释了α-CTLA-4对淋巴结和肿瘤中T eff和T reg细胞积聚的矛盾影响。更重要的是,他们强调了肿瘤微环境在决定基于抗体的免疫治疗结果方面发挥的重要作用,以及对细胞隔室的影响在肿瘤周边和肿瘤中如何不同。最后,他们建议利用肿瘤微环境的能力耗尽抗体相关T调节细胞的方法可以用于增强免疫治疗的抗肿瘤活性。
结果
GVAX+α-CTLA-4联合治疗通过CD4依赖机制保护B16-BL6黑色素瘤
确定CD4的参与+肿瘤保护中的T细胞室、C57BL/6野生型和I-A−/−小鼠(缺乏CD4+T细胞室)用可移植的B16-BL6黑色素瘤细胞系进行挑战。植入后第3天,用照射过的B16-BL6肿瘤细胞疫苗治疗或不治疗小鼠,该疫苗在存在或不存在阻断CTLA-4(α-CTLA-4)的单克隆抗体的情况下分泌GM-CSF(GVAX)。与之前的工作保持一致(van Elsas等人,1999年)GVAX和α-CTLA-4的联合使用可以保护小鼠免受肿瘤生长的影响,而单用每种药物都不能(图1 A). I-A失去保护−/−老鼠(图1、A和B),表明CD4+T细胞对肿瘤排斥反应至关重要。与之前的工作一致(Quezada等人,2006年,2010;Liakou等人,2008年;Waitz等人,2012年),α-CTLA-4显著增强肿瘤内CD4+Foxp3系列−/CD4细胞+Foxp3系列+(T eff/T reg cell)比率(图1 C).
图1。
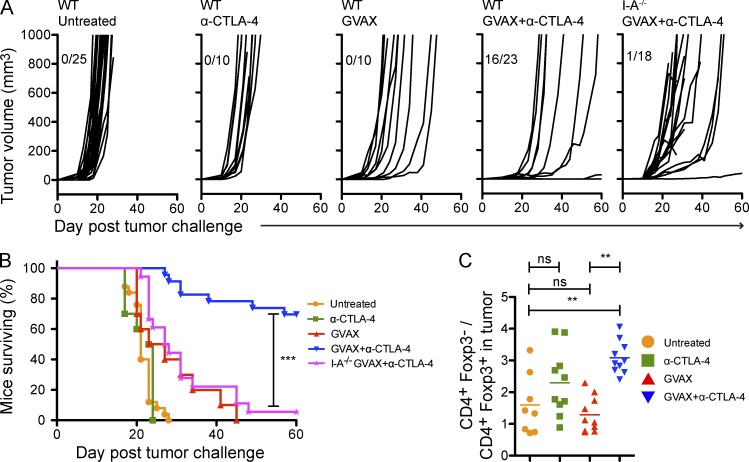
GVAX+α-CTLA-4联合治疗通过CD4保护肿瘤生长+T细胞依赖机制。(A) 小鼠受到10次挑战4B16-BL6在第0天,然后在第3天、第6天和第8天不治疗或使用GVAX、α-CTLA-4或GVAX+α-CTLA-4治疗。结果显示了单个小鼠的肿瘤生长曲线(A)和累积存活率(B)。n个=每组10–25只小鼠,死亡事件对应于肿瘤体积>350 mm三或者老鼠的死亡。(C) 用5×10攻击小鼠4B16-BL6,并按上述方法进行处理。CD4+Foxp3系列−/CD4细胞+Foxp3系列+计算淋巴结和肿瘤的比值。n个=每组8-10只小鼠。**,P<0.01;***,P<0.001。实验重复三次并合并。
α-CTLA-4增加肿瘤特异性CD4的数量+淋巴结中的T效率和T调节细胞,但可防止肿瘤内T调节细胞积聚
鉴于T eff/T reg细胞比率被认为会影响免疫治疗的结果(Quezada等人,2011年),我们重点研究了调节T eff和T reg细胞数量以响应α-CTLA-4的机制。追踪肿瘤/自身抗原特异性CD4+T细胞,我们使用了一种最近开发的TCR转基因小鼠,它可以产生CD4+I-A上呈现的黑素细胞分化抗原Trp1/gp75特异性T细胞b条(以下简称Trp1 T细胞;Muranski等人,2008年). 大约10%的Trp1 T细胞表达调节转录因子Foxp3(图2 A),之前的研究已经证明Trp1Foxp3−细胞(Trp1T-eff细胞)促进自身免疫性白癜风的发展,而Trp1Foxp3+细胞(Trp1T调节细胞)抑制它(谢等人,2010).
图2。
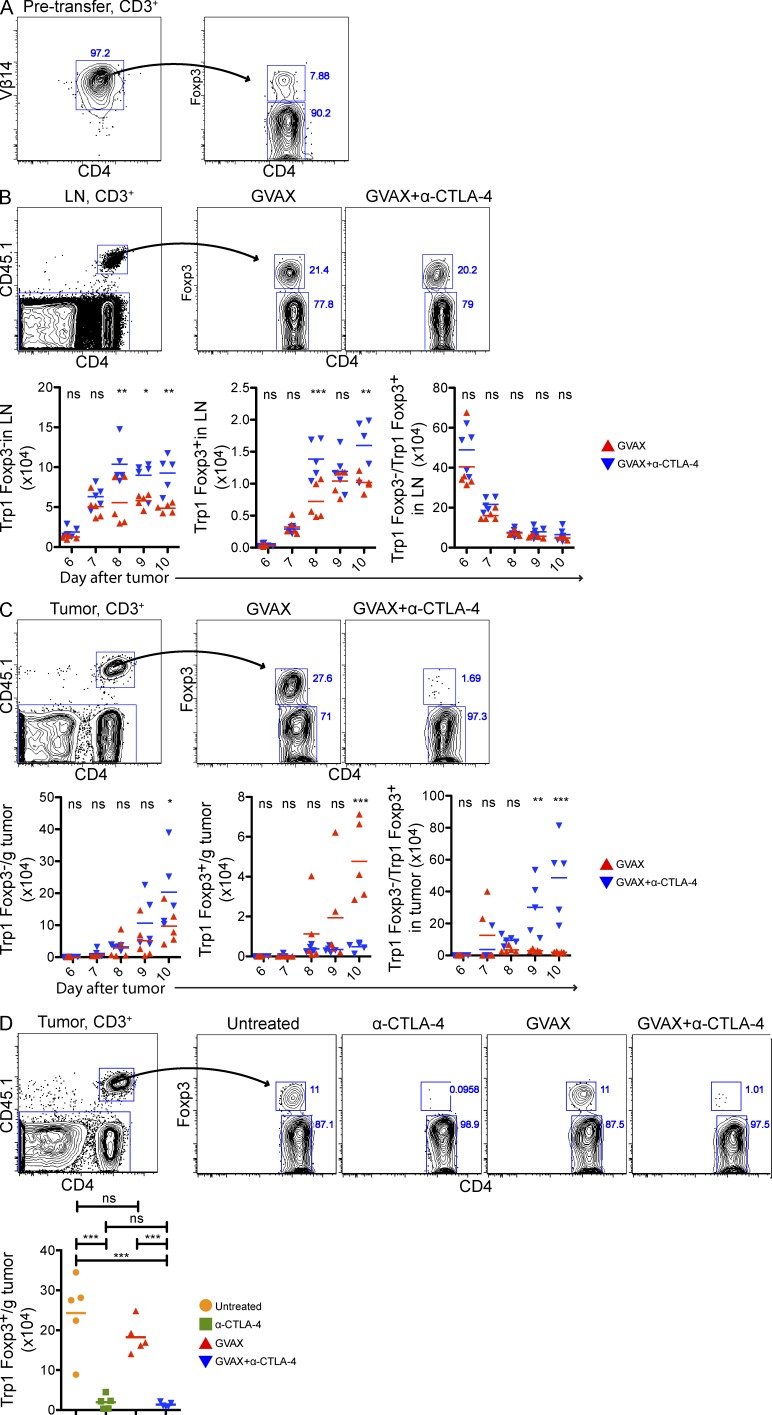
α-CTLA-4增加肿瘤特异性CD4的数量+淋巴结中的T效率和T调节细胞,同时防止肿瘤内T调节细胞积聚。用1.5×10攻击小鼠5B16-BL6第0天,用5×10过户4CD4细胞+CD45.1型+Trp1 T在第3天,并在第3、6和8天用GVAX或GVAX+α-CTLA-4治疗。(A) CD4表达Foxp3+Vβ14+转移前从Trp1 SJL RAG tan小鼠分离的细胞。CD4的(B和C)绝对细胞数和Foxp3表达+CD45.1型+来自淋巴结(B)和肿瘤(C)的Trp1 T细胞。(D) 肿瘤内CD4表达Foxp3+CD45.1型+Trp1 T细胞。小鼠如上所述进行治疗,但也未经治疗或用α-CTLA-4单药治疗。n个=每组5只小鼠。*,P<0.05;**,P<0.01;***,P<0.001。实验重复了两次。
B16-BL6植入后3天,小鼠接受50000 CD45.1+Trp1 T细胞与GVAX或GVAX和α-CTLA-4联合静脉注射。使用CD45.1标记物定量Trp1的积累。在实验过程中,GVAX处理小鼠淋巴结中Trp1T eff和Trp1T-reg细胞均蓄积(图2 B). 然而,Trp1T调节细胞优先积累(与高水平增殖相关抗原Ki67的表达相关;未发表的数据),导致T效率/T调节细胞比率降低。α-CTLA-4增加了两个群体的绝对数量,但没有改变Trp1T eff细胞/Trp1T reg细胞比率(图2 B). 相反,尽管GVAX治疗小鼠的肿瘤中积累了Trp1T-eff细胞和Trp1T reg细胞,但α-CTLA-4显著减少了Trp1 T reg的细胞数量,显著增加了肿瘤内Trp1T-eff细胞/Trp1T-reg细胞的比率(图2 C). 重要的是,α-CTLA-4作为单一疗法也减少了肿瘤内Trp1T调节细胞的积累(图2 D)表明肿瘤内T调节细胞数量的减少并不依赖于GVAX的给药。三个不同的α-CTLA-4克隆将肿瘤浸润Trp1T调节细胞的数量降低到不同的水平(图3),这表明这些结果可能不仅仅依赖于α–CTLA-4单克隆抗体阻断CTLA-4的能力。
图3。
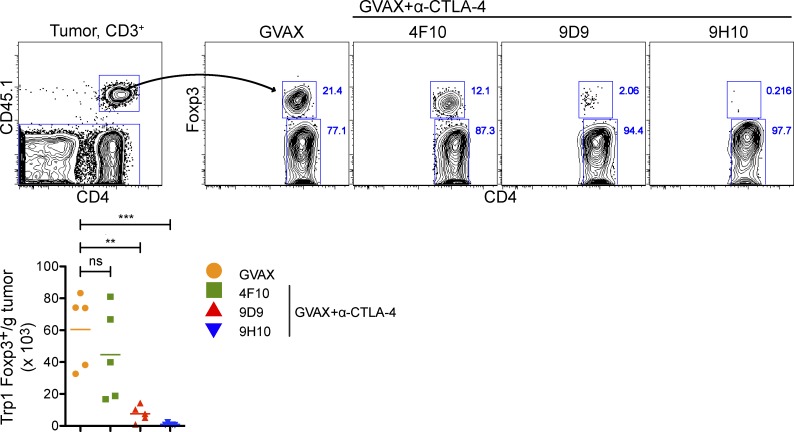
多个α–CTLA-4克隆减少了肿瘤内Trp1 T调节细胞的数量。按照图2带有α-CTLA-4克隆4F10、9D9或9H10。结果显示肿瘤内CD4的绝对细胞数和Foxp3表达+CD45.1型+Trp1 T细胞。n个=每组5只小鼠。*,P<0.05;**,P<0.01;***,P<0.001。实验重复两次。
α-CTLA-4诱导肿瘤浸润性CTLA-4快速消除+T调节细胞
为了了解α-CTLA-4治疗后肿瘤内Trp1T调节细胞数量的调节机制,我们评估了Trp1Treg细胞清除的动力学。用GVAX单药治疗小鼠至第10天,以使Trp1T调节细胞浸润强劲(图4 A). 在整个实验过程中,或在第10天用α-CTLA-4单剂量处理小鼠,然后在1天后处死。令人惊讶的是,这两种治疗都显著减少了肿瘤浸润Trp1T调节细胞的数量(图4 A)表明耗竭是一种潜在机制。
图4。
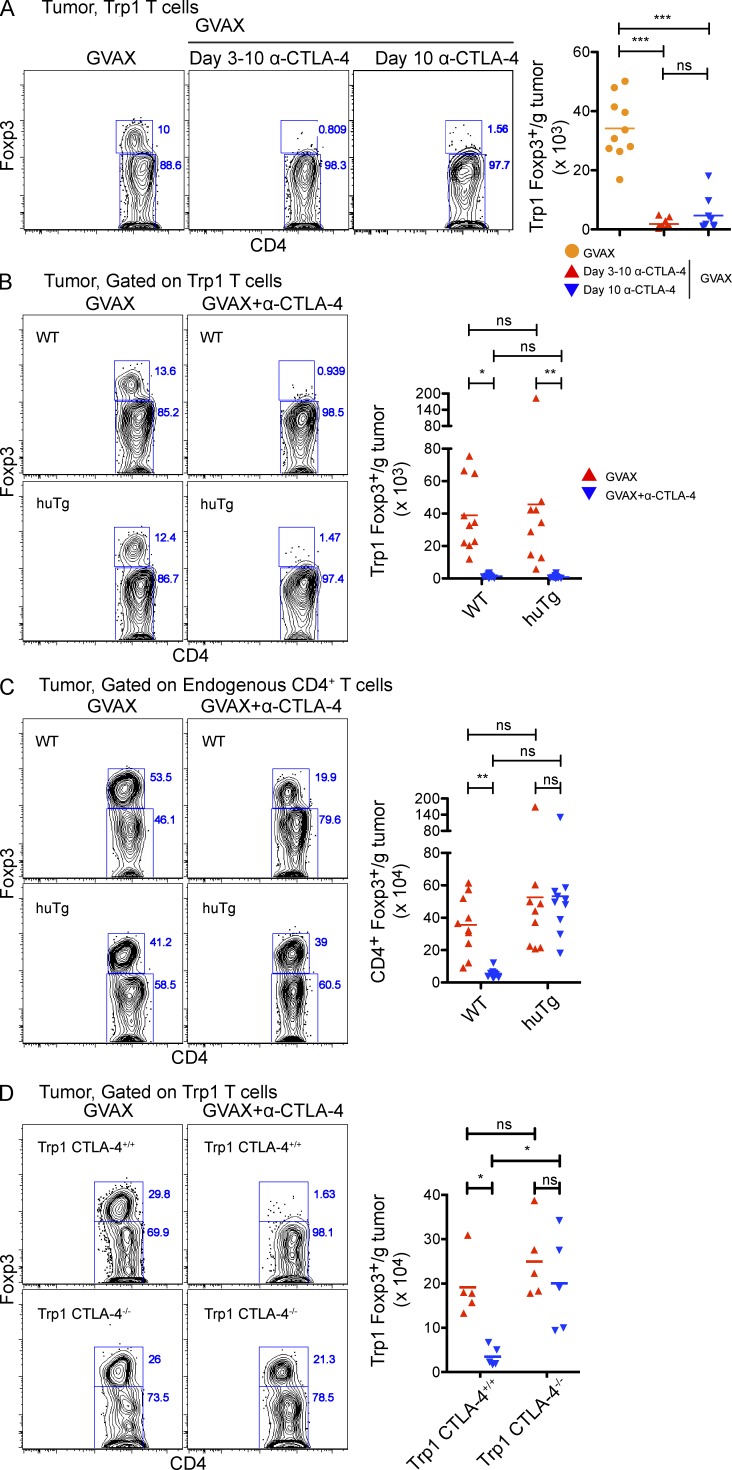
肿瘤特异性T reg细胞通过α-CTLA-4快速且自主地从肿瘤中清除。(A) 用1.5×10攻击小鼠5B16-BL6第0天,用5×10过户4CD4细胞+CD45.1型+Trp1 T在第3天,并在第3、6和8天用GVAX或GVAX+α-CTLA-4处理。一些小鼠没有接受进一步治疗,或在第3-10天或仅在第10天接受α-CTLA-4治疗,然后在第11天处死。结果显示肿瘤内CD4的绝对细胞数和Foxp3表达+CD45.1型+Trp1 T细胞。n个=每组9-10只小鼠。(B和C)野生型和CTLA-4-huTg小鼠按照图2结果显示肿瘤内CD4的绝对细胞数和Foxp3表达+CD45.1型+Trp1(B)和CD4+CD45.1型−内源性多克隆T细胞(C)。(D) CTLA-4型+/+和CTLA-4−/−将Trp1 T细胞转移到野生型小鼠中,然后按照图2结果显示肿瘤内CD4的绝对细胞数和Foxp3表达+CD45.1型+Trp1 T细胞。*,P<0.05;**,P<0.01;***,P<0.001。实验重复两次并合并。
为了研究α–CTLA-4单克隆抗体与Trp1T调节细胞的直接结合是否介导耗竭,我们将Trp1 T细胞转移到表达人而非小鼠CTLA-4(CTLA-4-huTg;Peggs等人,2009年). 用GVAX和抗小鼠CTLA-4(9H10克隆不与人CTLA-4结合)治疗后,Trp1T reg细胞表达小鼠CTLA4(图4 B),但不是内源性多克隆T调节细胞-表达人CTLA-4(图4C)α-CTLA-4抗体和CTLA-4之间的直接相互作用+Trp1T调节细胞需要介导肿瘤的耗竭。
为了证实α-CTLA-4治疗是否通过直接结合和耗尽表达CTLA-4的T调节细胞而不是阻断CTLA-4功能来消除肿瘤浸润的Trp1T调节细胞,我们采用了从CTLA-4转移Trp1 T细胞的方法+/+或CTLA-4−/−Trp1 TCR转基因动物,然后测量肿瘤内Trp1T调节细胞对GVAX或GVAX和α-CTLA-4的反应。而α-CTLA-4的加入导致肿瘤浸润CTLA-4的数量显著减少+/+Trp1 T调节细胞,肿瘤浸润CTLA-4的数量无变化−/−α-CTLA-4治疗后观察到Trp1细胞(图4 D). 这些数据表明,肿瘤中肿瘤反应性T reg细胞数量的减少并不依赖于“阻断”CTLA-4介导的调节,因为肿瘤反应性Trp1细胞上的CTLA-4基因消融并没有导致肿瘤中Trp1T reg的清除。
CTLA-4的快速消除+Trp1T reg细胞支持耗竭作为潜在机制,尽管有其他机制,如Foxp3表达缺失+单元格(Zhou等人,2009年),或Foxp3分化减少−到Foxp3+细胞(转化;Benigni等人,2006年;Moo-Young等人,2009年)也可以解释Trp1T调节细胞的减少。为了测试这些可能性,我们使用了Trp1Foxp3GFP公司报告小鼠在Foxp3启动子下表达GFP以测量转化,并同源标记(CD45.1和/或CD45.2)Trp1T eff细胞(Foxp3−)和Trp1T调节细胞(Foxp3+)用于跟踪治疗反应中每个隔室的命运的子集。Trp1GFP基因−(Foxp3−)转移到未经治疗或接受GVAX、α-CTLA-4或GVAX+α-CTLA-4治疗的荷瘤小鼠体内的细胞未能上调GFP或Foxp3(图5 A),表示Foxp3−到Foxp3+转化并没有促进肿瘤内Trp1T reg细胞的积累。因此,这些数据还表明,α-CTLA-4并不能通过阻止转化来消除肿瘤中的Trp1T调节细胞。为了测试T调节细胞室中Foxp3表达的潜在损失,小鼠被给予CD45.1的混合物+/+CD25型−Trp1T-eff细胞和CD45.1+/−CD45.2型+/−CD25型+Trp1T调节细胞,然后用GVAX或GVAX+α-CTLA-4治疗(图5 B). 在GVAX+α-CTLA-4治疗后,CD45.1+/−CD45.2型+/−CD25型+从肿瘤中完全消除Trp1T调节细胞群,以防止Foxp3表达的丢失(图5 B). 此外,在T调节细胞(未显示)中未观察到增殖标记Ki-67的表达存在显著差异,但这是肿瘤浸润T调节细胞丢失的潜在机制。这些数据支持一种模型,即α-CTLA-4通过消耗而不是通过T reg细胞增殖的改变或Foxp3表达的丧失从肿瘤中消除Trp1T reg。
图5。
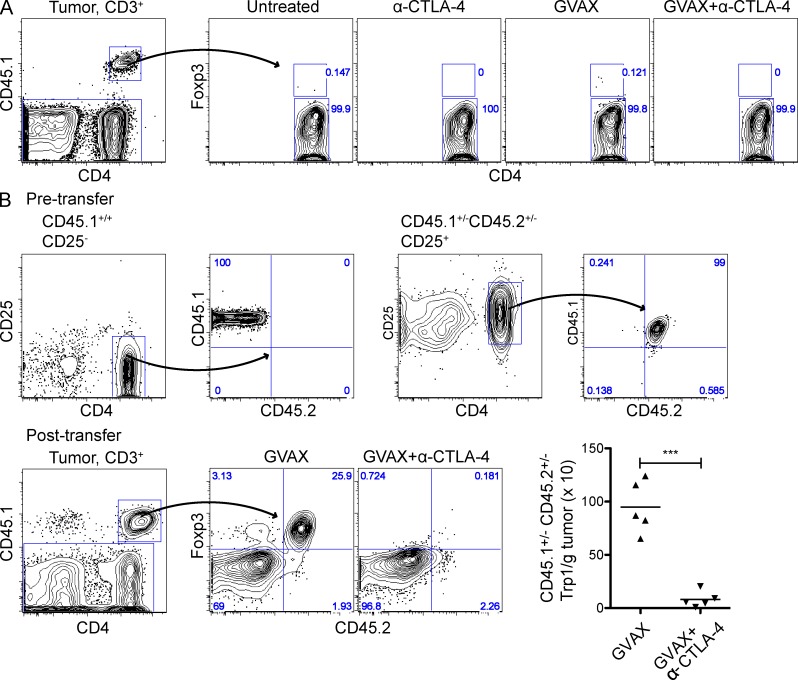
肿瘤特异性T调节细胞被α-CTLA-4从肿瘤中清除。(A) CD4细胞+GFP公司−细胞是从CD45.1中纯化的FACS+/+B类W公司RAG公司−/−Trp1-Foxp3-GFP小鼠,过继性转移到具有三维已建立肿瘤的小鼠中,并按照图2结果显示CD4表达Foxp3+CD45.1型+肿瘤中的Trp1细胞。(B) 用1.5×10攻击小鼠5第0天的B16-BL6,CD25的混合物−CD45.1型+/+和CD25+CD45.1型+/−CD45.2型+/−在第3天过继转移Trp1 T细胞,然后按照图2.(顶部)分选CD25的表型−和CD25+混合和转移前的Trp1 T细胞。(底部)肿瘤内CD4的绝对细胞数和CD45.2表达+CD45.1型+Trp1 T细胞。n个=每组5只小鼠。*,P<0.05;**,P<0.01;***,P<0.001。实验重复了两次。
T调节细胞耗竭主要由FcγRIV介导
抗体介导的消耗可通过补体介导的裂解发生(Reff等人,1994年;Giorgini等人,2008年;Wang和Weiner,2008年)或抗体依赖性细胞介导的细胞毒性(ADCC;Swanson和Hoppe,2004年;斯图亚特和埃泽科维茨,2005年;Nimmerjahn和Ravetch,2008年). 我们首先使用补体缺乏的C3−/−老鼠(Giorgini等人,2008年)作为肿瘤反应性Trp1 T细胞的受体,仍然观察到肿瘤浸润性Trp1T调节细胞对α-CTLA-4的反应耗尽(图6 A). 因此,我们调查了ADCC在我们的系统中的潜在参与。在小鼠中,ADCC主要由低亲和力受体FcγRIII和FcγRIV调节(Nimmerjahn等人,2005年;Nimmerjahn等人,2010年;Setiady等人,2010年),在较小程度上由高亲和力FcγRI(Fossati-Jimack等人,2000年;Tedder等人,2006年;Hamaguchi等人,2006年). 为了确定FcγRs是否介导T reg细胞耗竭,我们使用γ−/−缺乏所有三种激活FcγRs的连锁小鼠作为受体(Takai等人,1994年;Clynes等人,1998年). 虽然Trp1T调节细胞(图7 A)和内源性多克隆T调节细胞亚群(图7 B)经α-CTLA-4治疗的野生型小鼠的肿瘤显著减少,γ−/−链受体小鼠接受α-CTLA-4治疗,表明通过FcγR依赖机制耗竭。消耗也独立于FasL和TRAIL细胞毒性途径发生(图6 B).
图6。
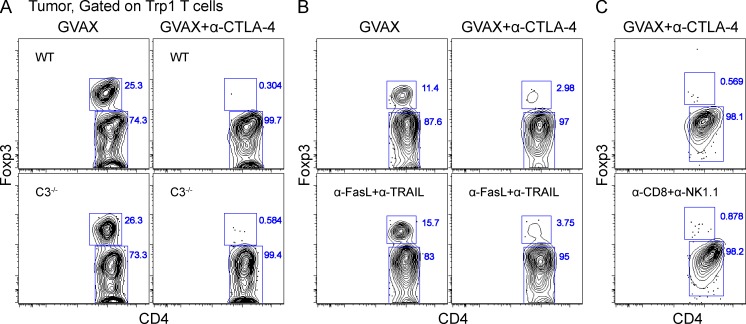
肿瘤特异性T调节细胞不会被补体、FasL–TRAIL途径或NK/CD8耗尽+T细胞。(A–C)按照图2结果显示CD4表达Foxp3+CD45.1型+补体C3中的Trp1细胞−/−小鼠(A)、用阻断α-FasL和α-TRAIL抗体治疗的小鼠(B)或用耗尽α-CD8和α-NK1.1抗体治疗的鼠(C)。实验重复了两次。
图7。
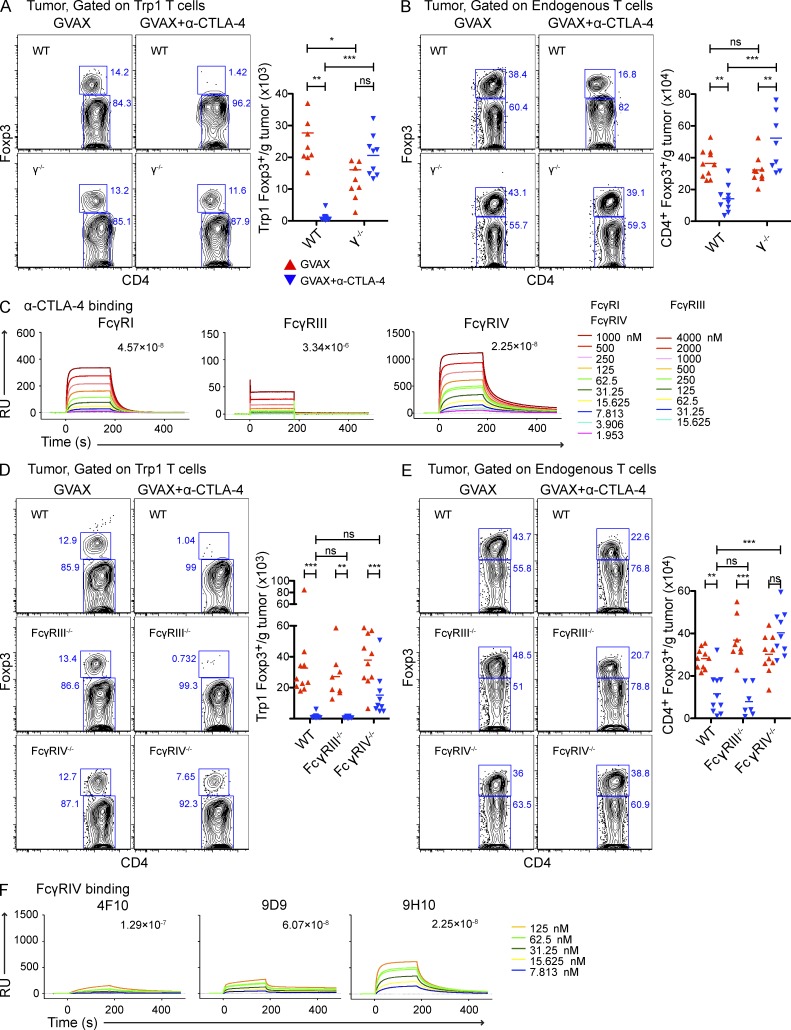
T调节细胞耗竭主要由FcγRIV介导。(A和B)野生型和γ−/−用B16-BL6攻击小鼠,并按照图2肿瘤内CD4的绝对细胞数和Foxp3表达+CD45.1型+Trp1(A)和CD4+CD45.1型−内源性多克隆T细胞(B)。n个=每组9-10只小鼠。(C) α-CTLA-4克隆9H10与FcγRI、FcγRAII和FcγROV结合的SPR分析。(D和E)野生型,FcγREII−/−和FcγRIV−/−用B16-BL6攻击小鼠,并按照图2肿瘤内CD4的绝对细胞数和Foxp3表达+CD45.1型+Trp1(D)和CD4+CD45.1型−内源性多克隆T细胞(E)。(F) 结合FcγRIV的三个不同α–CTLA-4克隆(4F10、9D9、9H10)的SPR分析。n个=每组7–10只小鼠。*,P<0.05;**,P<0.01;***,P<0.001。实验重复三次并合并。
利用表面等离子体共振(SPR),我们确定了α–CTLA-4与不同FcγRs的结合。α-CTLA-4克隆9H10与FcγRI结合的亲和力最高(图7 C). 在ADCC中涉及的两个低亲和力FcγRs中,9H10与FcγRIV结合的亲和力较高,表明其更容易参与衰竭。当我们比较野生型和FcγRIII中α-CTLA-4对肿瘤浸润性T调节细胞的体内耗竭时−/−和FcγRIV−/−小鼠,我们观察到FcγRIV中Trp1T调节细胞数部分恢复,内源性多克隆T调节细胞数量完全恢复−/−,但不在FcγRIII中−/−老鼠(图7、D和E)支持FcγRIV在α-CTLA-4治疗后肿瘤浸润性T调节细胞耗竭中的主要作用。有趣的是,我们的三个不同α–CTLA-4克隆与FcγRIV的结合亲和力(图7 F)与体内清除肿瘤浸润T调节细胞的能力相关(图4 A). 表达FcγRIII但无其他激活FcγRs的NK细胞耗竭(Takai等人,1994年;Nimmerjahn和Ravetch,2008年),不能阻止Trp1T调节细胞对α-CTLA-4的反应(图6 C),进一步支持FcγRIII在该模型中T调节细胞耗竭中的不必要性。然而,FcγRIV中Trp1T调节细胞的部分恢复−/−与内源性多克隆T调节细胞室的完全恢复相比,小鼠表明FcγR系统中存在潜在冗余。这可以通过靶分子表达水平的差异来解释,因为Trp1 T调节细胞表达的表面CTLA-4水平高于内源性T调节细胞(参见图9、A和B),可能增加其对其他FcγRs耗竭的敏感性,以补偿FcγRIV的损失。与此相一致,FcγR与9H10克隆具有高亲和力(图7 C),也能诱导ADCC(Otten等人,2008年),提示FcγR在Trp1T调节细胞耗竭中的潜在作用。尽管如此,我们的数据表明FcγRIV是内源性肿瘤浸润T调节细胞耗竭的关键介质。
图9。
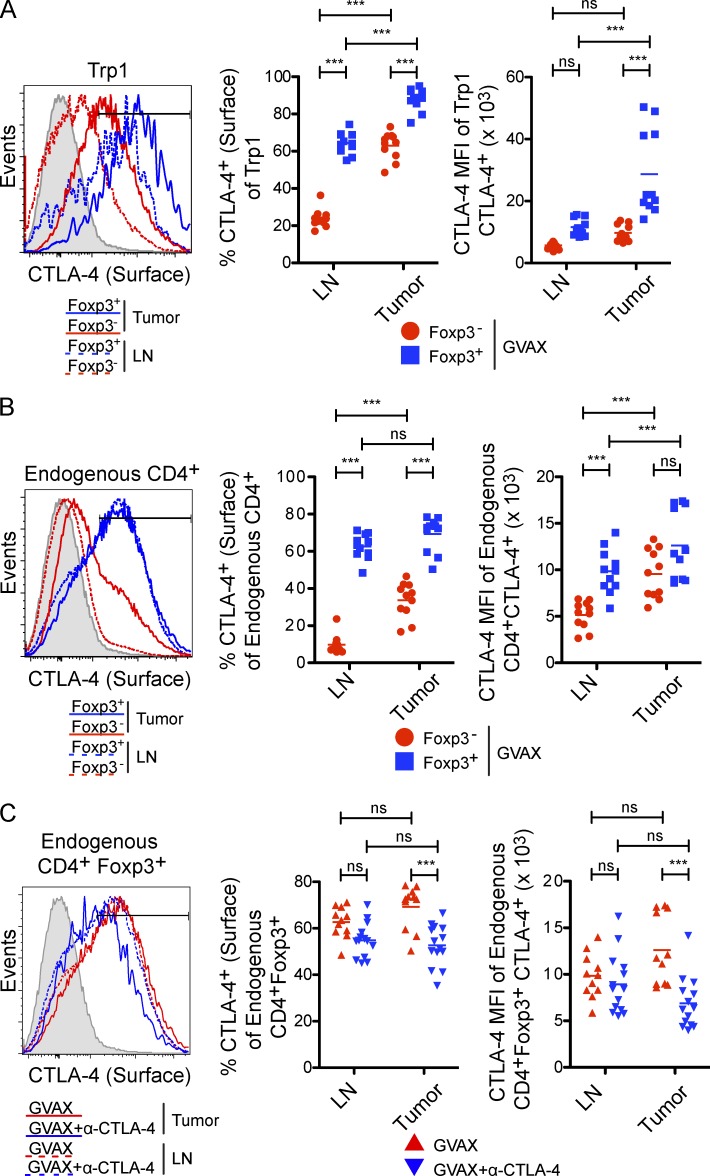
肿瘤浸润性T reg细胞表达高水平的表面CTLA-4。CD4表达(A和B)表面CTLA-4+CD45.1型+Trp1(A)和CD4+CD45.1型−GVAX治疗小鼠肿瘤和淋巴结中的内源性多克隆T细胞(B)(如图2). 阴影直方图表示CTLA-4-huTg小鼠。n个=每组11只小鼠。(C) CD4表面CTLA-4表达的定量+Foxp3系列+GVAX或GVAX+α-CTLA-4治疗后淋巴结和肿瘤中的多克隆T调节细胞。小鼠接受GVAX或GVAX+α-CTLA-4(克隆9H10)治疗,如图2并在第10天献祭。内源性CD4表面CTLA-4的表达+Foxp3系列+通过α-CTLA-4克隆4F10染色检测T调节细胞,该克隆未被克隆9H10交叉阻断。n个=每组11–14只小鼠。*,P<0.05;**,P<0.01;***,P<0.001。实验重复两次并合并。
CD11b型+FcγRIV+巨噬细胞在肿瘤微环境中富集
考虑到小鼠的ADCC主要由巨噬细胞介导(Uchida等人,2004年;Tedder等人,2006年;Setiady等人,2010年)接下来,我们评估了肿瘤内巨噬细胞和其他免疫亚群上FcγRIV的表达。如前所示(Nimmerjahn等人,2005年),FcγRIV的表达仅限于CD11b+单元格(图8 A). 赖氨酸6C你好赖氨酸6G−巨噬细胞(Shi等人,2011年)构成CD11b的大多数+FcγRIV+种群,而Ly6洛赖氨酸6G+中性粒细胞和CD11c+I-A公司+树突细胞只是一个次要成分(图8 B). 特别值得注意的是,CD11b+相对于淋巴结,肿瘤中的细胞富集了70倍(图8 C),其中未发生消耗(图2 B). 巨噬细胞特异性标记物Iba1的染色组织切片证实肿瘤中存在巨噬细胞(Graeber等人,1998年)与淋巴结相比,肿瘤中的巨噬细胞密度要高得多(图8 D). 最后,肿瘤内CD11b上FcγRIV的表达显著升高+细胞,但在CD11b上几乎检测不到+淋巴结细胞(图8 E). 重要的是,α-CTLA-4没有改变CD11b的百分比+肿瘤中的细胞(图8 C)或通过肿瘤浸润CD11b表达FcγRIV+单元格(图8 E),这表明T reg细胞耗竭并非由于肿瘤中巨噬细胞对α-CTLA-4的反应数量或功能普遍增加所致。
图8。
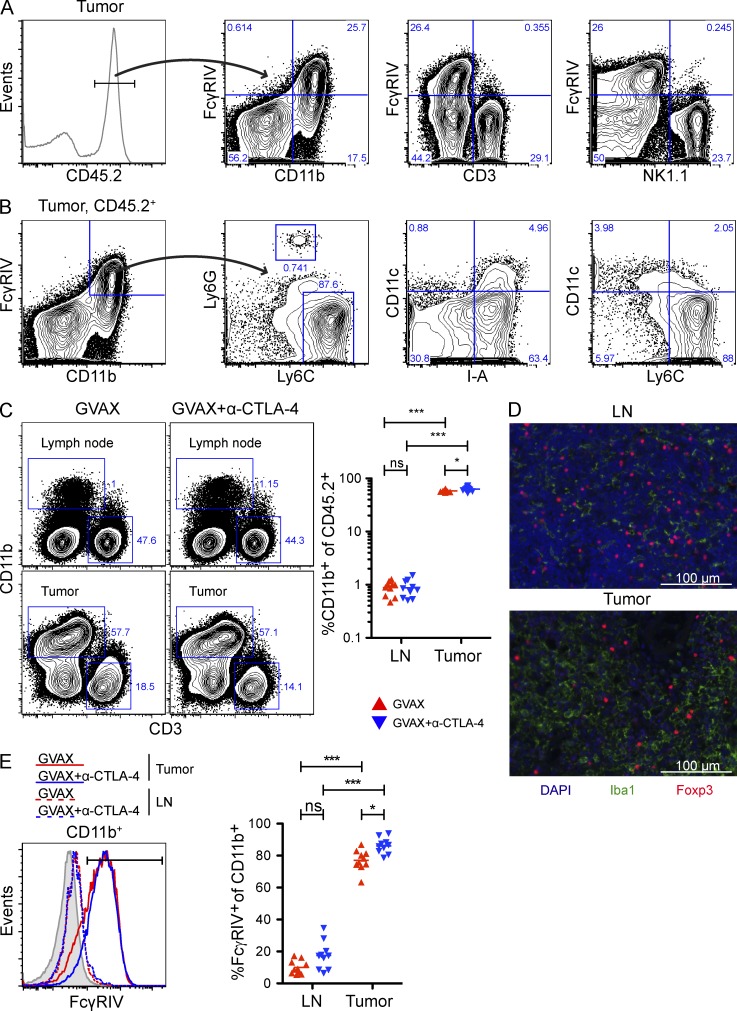
表达高水平FcγRIV的吞噬细胞在肿瘤微环境中富集。(A–E)按照图2(A)肿瘤内CD11b代表性FcγRIV表达+,NK1.1+和CD3+细胞。(B) 肿瘤内CD45.2的代表性CD11c、I-A、Ly6C和Ly6G表达+CD11b型+FcγRIV+细胞。(C) CD45.2上CD3和CD11b的表达+淋巴结和肿瘤中的细胞。n个=每组10只小鼠。(D) GVAX治疗小鼠淋巴和肿瘤中Iba1和Foxp3的表达。棒材,100µm。(E) CD11b表达FcγRIV+淋巴结和肿瘤中的细胞。阴影直方图表示CD11b的FcγRIV表达+FcγRIV细胞−/−老鼠。n个=每组10只小鼠。*,P<0.05;***,P<0.001。实验重复两次并合并。
肿瘤浸润性T调节细胞表面CTLA-4表达升高
α-CTLA-4对肿瘤内Trp1T调节细胞和内源性T调节细胞的选择性耗竭(图2 C和7亿)表明α-CTLA-4优先与T调节细胞室相关。与此一致,CTLA-4在Trp1T调节细胞上的表面表达显著升高(图9 A)和内源性T调节细胞(图9 B)与对应的效应器相比。此外,与内源性T调节细胞相比,Trp1T调节细胞表达更高水平的表面CTLA-4(图9、A和B)并且更容易被α-CTLA-4耗尽(图7,A和B). 此外,与淋巴结相比,肿瘤中表面CTLA-4的表达增加,而淋巴结中没有出现缺失(图2、B和C). 最后,仅在α-CTLA-4治疗后肿瘤部位T调节细胞室表面CTLA-4水平显著降低(图9 C). 这些数据表明,肿瘤中T调节细胞的缺失与T调节细胞亚群增加的CTLA-4表面表达和CD11b的富集有关+FcγRIV+肿瘤微环境中的巨噬细胞。
α–CTLA-4治疗不会增加FcγRIV的瘤内T eff/T reg细胞比率−/−小鼠,并且未能诱导肿瘤保护
为了确定FcγRIV表达缺失和肿瘤内T调节细胞缺失是否影响肿瘤保护,我们评估了WT和FcγRAV中GVAX和α-CTLA-4的抗肿瘤活性−/−老鼠。在野生型小鼠中,α-CTLA-4显著增加了T eff/T reg细胞(Trp1和内源性)的瘤内比率,而在FcγRIV中却没有这样做−/−老鼠(图10,A和B),表明FcγRIV对T reg细胞的耗竭显著有助于肿瘤内T eff相对于T reg细胞的相对增加。重要的是,FcγRIV−/−经GVAX和α-CTLA-4治疗后,小鼠也未能排斥B16-BL6黑色素瘤。(图10 C)支持α-CTLA-4的消耗活性与其抗肿瘤活性的相关性。总之,我们的数据说明了肿瘤微环境的特定特征在介导T调节细胞耗竭和确定α-CTLA-4治疗后的结果方面的关键重要性。
图10。
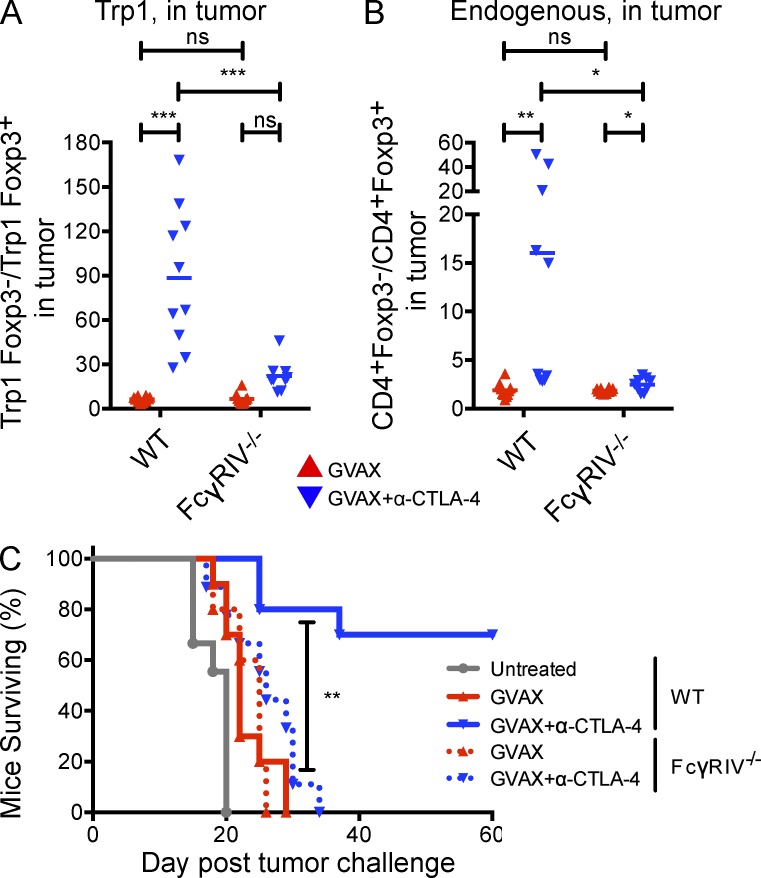
α-CTLA-4治疗不会增加FcγRIV中肿瘤内T eff/T-reg细胞比率−/−而未能诱导肿瘤保护。(A和B)CD4的T eff/T reg细胞比率+CD45.1+Trp1(A)和CD4+肿瘤中的CD45.1−内源性多克隆T细胞(B)。n个=每组9-10只小鼠。(C) 野生型和FcγRIV的存活率−/−用GVAX或GVAX+α-CTLA-4治疗的小鼠,如图1 A.n个=每组10–11只小鼠。*,P<0.05;***,P<0.001。实验重复两次并合并。
讨论
α-CTLA-4治疗在临床前肿瘤模型中显示出显著的抗肿瘤活性(Leach等人,1996年;van Elsas等人,1999年;Quezada等人,2011年)以及作为转移性黑色素瘤免疫治疗的临床试验(Hodi等人,2010年;Robert等人,2011年). 在动物模型和临床中,我们和其他人已经证明α-CTLA-4增加了肿瘤内CD8+/T调节细胞和T eff/T调节细胞比率(图2 C;Quezada等人,2006年;Hodi等人,2008年;Liakou等人,2008年),考虑到T eff和T reg细胞的抗肿瘤和促肿瘤生长活性,它被认为会驱动肿瘤排斥反应(Griffiths等人,2007年;Quezada等人,2008年).
在这里,我们进一步探讨了α-CTLA-4治疗增加肿瘤内T eff/T reg细胞比率的机制,重点是对T reg细胞室的影响。我们证明,α-CTLA-4治疗通过FcγR依赖机制消耗T调节细胞,从而增加了这一比率。α–CTLA-4介导的T reg细胞耗竭的显著区室化揭示了α–CTLA-4 mAb功能的一个先前意想不到的特征,这一点很重要,原因有几个。首先,它可能有助于定义临床有效单克隆抗体的特定治疗相关属性,允许将这些特征工程到第二代抗体中,并将其应用于临床。其次,微环境在促进这种α-CTLA-4活性方面的关键重要性可能有助于开发合理的组合疗法,旨在优化与最大α-CTLA-4介导的耗竭相关的微环境线索,即。,肿瘤浸润抗原呈递细胞/巨噬细胞上FcγR表达的特定模式。第三,评估肿瘤微环境的这些特征可能为预测α-CTLA-4治疗的反应性提供有价值的生物标记物。基于T reg细胞亚群在抑制多种恶性肿瘤的抗肿瘤免疫反应中的重要性(Antony等人,2005年;Chen等人,2007年;李和叶,2008;Quezada等人,2008年;Klages等人,2010年),我们预测,含有少量巨噬细胞或表达低水平FcγRs的巨噬细胞的肿瘤对α-CTLA-4治疗的反应较差,而增强此类细胞对肿瘤浸润的治疗可能会协同作用,使其成为进一步临床评估的理想候选。值得注意的是,在临床环境中,肿瘤内NK细胞的丰度也可能影响α-CTLA-4的活性,因为人类NK细胞表达FcγRIIIA,这是小鼠中FcγRIV的同源物,也是ADCC的主要介质(Nimmerjahn和Ravetch,2008年).
FcγRIV基因消融部分恢复肿瘤特异性Trp1 T调节细胞,并完全恢复浸润肿瘤的内源性多克隆T调节细胞(图7、D和E),表明FcγR在耗竭中的主要作用。此外,最近的研究表明FcγRIV的基因缺失会导致体内ADCC的完全丧失(Nimmerjahn等人,2010年)进一步支持FcγRIV在肿瘤浸润性T调节细胞对α–CTLA-4反应的耗竭中的作用。FcγRIV的表达仅限于CD11b+肿瘤内的细胞(图8 A)主要是Ly6C你好赖氨酸6G−(图8 B). 巨噬细胞对T调节细胞的消耗与之前的工作一致,表明在小鼠中,ADCC主要由该免疫亚群介导(Stockmeyer等人,2001年;Uchida等人,2004年;Tedder等人,2006年;Oflazoglu等人,2007年)NK细胞发挥更为有限的作用,这是由单个FcγR、FcγRIII的表达引起的(Nimmerjahn和Ravetch,2008年). 与此模型一致,肿瘤中的T调节细胞被耗尽(图2 C)、派尔氏贴片和疫苗位点(未描述),其中含有大量CD11b+单元格(图8 C但不是来自淋巴结(图2 B)和脾脏(未描绘),其中含有少量CD11b+FcγRIV水平几乎检测不到的细胞(图8 C). 虽然T调节细胞耗竭与CD11b的存在之间存在关联+FcγRIV+肿瘤中的细胞看起来很清楚,GVAX对CD11b的高频率的额外贡献+FcγRIV+肿瘤中发现的细胞(图8,C–E)需要进一步评估,因为已知GVAX分泌的GM-CSF有助于炎症巨噬细胞表型(Stockmeyer等人,2001年). 重要的是,肿瘤反应性T调节细胞的耗竭依赖于α-CTLA-4的给药,而不是GVAX(图2 D). 虽然我们的数据支持α-CTLA-4介导肿瘤浸润性T调节细胞的耗竭这一观点,但它也表明,对于免疫原性较差的肿瘤(如B16模型),GVAX或其他组合方法可能有必要增加肿瘤和淋巴结中肿瘤反应性效应T细胞的频率(图2、B和C). 添加α-CTLA-4将进一步增加LN和肿瘤中肿瘤反应性效应T细胞的数量(图2、B和C)同时消耗肿瘤浸润的T调节细胞(图2 C、2 D).
最后,我们最近评估了α–CTLA-4对已知对α–CTLA-4单药治疗有反应的结肠癌(CT26)小鼠模型中肿瘤浸润T调节细胞的影响。在这个模型中,我们还观察到α-CTLA-4作为单一药物对肿瘤浸润性T调节细胞的反应显著减少。此外,CT26模型中肿瘤浸润性T reg细胞的减少也仅限于肿瘤部位,并与肿瘤内T eff/T reg细胞比率、肿瘤排斥反应和CD11b的肿瘤浸润性增加相关+FcγRIV+细胞(未发表的数据),与另一篇最近发表的结果一致,该结果表明抗体同型在CT26模型中α-CTLA-4抗肿瘤活性中的重要性(Selby等人,2013年).
我们的数据还表明,表达或诱导表达能够驱动ADCC的细胞亚群的肿瘤最有可能介导T reg细胞对α-CTLA-4的耗竭。在最近的III期临床试验中,α-CTLA-4与肽疫苗联合应用缺乏附加或协同治疗益处(Hodi等人,2010年)至少可以部分解释为缺乏疫苗介导的对适当辅助细胞群的影响。已有文献证明,伊普利穆单抗(抗人CTLA-4)单药治疗可显著降低膀胱癌患者肿瘤内T调节细胞的数量,这可能与该肿瘤类型固有的特定微环境有关(Liakou等人,2008年). 还需要注意的是,全人类α-CTLA-4单抗Ipilimumab与FcγRIIIA的结合亲和力(未公布的数据)与利妥昔单抗(抗CD20耗尽单抗)与FcβRIIIA之间的结合亲和力相当(神田等人,2007年),进一步支持临床活性α–CTLA-4单克隆抗体能够消耗T调节细胞的前提。显然,在α-CTLA-4治疗的临床应用中,无论是在记录肿瘤浸润性T调节细胞的潜在耗竭方面,还是在确定可能与ADCC介导的耗竭相关的人类恶性肿瘤中FcγR表达模式方面,都需要进行进一步的研究。
α-CTLA-4介导的T调节细胞耗竭的区域化,优先从含有较高CD11b的位点消除T调节细胞+细胞可能具有治疗优势。这种靶向活性可能会缓解肿瘤内的免疫抑制,同时使全身免疫基本保持完整。广泛消耗T调节细胞可诱发多种自身免疫性疾病(Fontenot等人,2003年;Kim等人,2007年). 然而,在使用伊普利单抗治疗的患者中,有一部分患者发生了与免疫相关的不良事件(Hodi等人,2010年;Robert等人,2011年). 有趣的是,结肠炎仍然是较常见的不良事件之一,而佩尔斑是我们的小鼠模型中T调节细胞耗竭的少数部位之一。进一步评估伊普利单抗对人类T调节细胞群的影响可能有助于解释潜在毒性,也有助于优化α-CTLA-4单抗的治疗相关特性。
我们之前证明CTLA-4与T eff细胞室的单室结合能够诱导一些小鼠的肿瘤保护(Peggs等人,2009年)尽管与靶向T eff和T reg细胞室相比,保护作用显著降低。相反,与T调节细胞室的单间室结合未能引起肿瘤保护,这突出了对T效应细胞室影响的重要性。这些结果现在可以用α-CTLA-4导致肿瘤微环境中的T调节细胞减少来解释。在目前的研究中,FcγRIV的保护作用被消融−/−老鼠(图10 C)不能消耗T调节细胞(图7 A和B),但应具有能够对α-CTLA-4治疗作出反应的T eff细胞室。这可能表明表达FcγRIV的细胞在介导T调节细胞耗竭以外的肿瘤保护中具有其他重要作用。或者,结果可能反映了α–CTLA-4对其他部位T调节细胞的影响,因为阻断可诱导淋巴结部位的增殖和积聚,而在单室T效应细胞阻断环境中不会发生。总的来说,这里提供的数据与α-CTLA-4免疫治疗释放T eff细胞室的细胞内抑制,同时靶向肿瘤微环境中的T reg细胞进行耗竭的模型一致。
我们的数据还提出了一个问题,即靶向其他共抑制或共刺激受体的抗体是否也通过肿瘤内T调节细胞缺失增强抗肿瘤免疫活性。α-OX40和α-GITR单克隆抗体均能诱导肿瘤微环境中T调节细胞的丢失(Coe等人,2010年;Hirschhorn-Cymerman等人,2012年)尽管机制仍不清楚。此外,这些抗体(例如,靶向T eff和T reg细胞室的α-OX40)的功能具有明显的双重性(Piconese等人,2008年)之前已注意到。
总之,我们的数据表明,α-CTLA-4单抗显著提高了肿瘤内T eff/T reg细胞比率,部分是通过依赖肿瘤微环境和特定单抗克隆的方式选择性地消耗T reg细胞。他们支持进一步评估利用肿瘤微环境耗尽抗体相关细胞的能力的临床策略,以及设计靶向T调节细胞优先高水平表达的共刺激和共抑制分子的耗尽抗体,优化以增强与特定肿瘤类型和靶向特定微环境最相关的FcγR的结合。
材料和方法
老鼠。
4-6周龄C57BL/6和I-A−/−小鼠购自英国杰克逊实验室和查尔斯河实验室。4-6周龄C57BL/6和γ−/−小鼠也从Taconic公司购买。FcγRIII−/−和FcγRIV−/−小鼠是从洛克菲勒大学(纽约,NY)的J.V.Ravetch获得的。之前描述的BW公司RAG公司−/−Trp1转基因小鼠(Muranski等人,2008年)由N.Restifo(马里兰州贝塞斯达国家癌症研究所)提供,并与B6.SJL小鼠杂交以产生CD45.1+/+B类W公司RAG公司−/−Trp1转基因小鼠。Trp1小鼠也与从A.Rudensky(纽约州纽约市斯隆-凯特琳纪念癌症中心)获得的Foxp3-GFP小鼠和CTLA-4小鼠杂交−/−小鼠产生CD45.1+/+B类W公司RAG公司−/−Trp1Foxp3型GFP公司和CD45.1+/+B类W公司RAG公司−/−Trp1CTLA-4型−/−分别是。小鼠在斯隆-凯特琳纪念癌症中心和英国查尔斯·里弗饲养。斯隆-凯特纪念癌症中心机构动物护理和使用委员会和伦敦大学学院动物伦理委员会批准了本手稿中的所有动物实验。
细胞系。
高致瘤性、低免疫原性B16-BL6细胞最初从I.J.Fidler(德克萨斯州休斯顿MD Anderson癌症中心)获得,并在完全RPMI(cRPMI)中培养。
抗体。
大多数阻断和中和抗体购自BioXCell,经静脉注射α-FasL(MFL4)和α-TRAIL(N2B2)抗体由H.Yagita(日本东京均田大学医学院)制备。9H10和4F10是仓鼠抗小鼠CTLA-4单抗,9D9是我们实验室产生的小鼠抗小鼠CTLA-4单抗。流式细胞仪和免疫荧光抗体购自eBioscience、BD和BioLegend。
Trp1 T细胞转移。
使用CO处死Trp1小鼠2安乐死。CD4细胞+使用CD4从脾脏和淋巴结(腹股沟、颈部、肠系膜、腋窝和肱骨)分离Trp1细胞+根据制造商的说明选择(Miltenyi Biotec),并通过尾静脉注射在PBS中过继转移。
肿瘤挑战和治疗。
在涉及Trp1 T细胞的功能实验中,用1.5×10的剂量对小鼠进行皮内激发(i.d.)5B16-BL6在第0天的右翼。肿瘤植入后3d,5×104CD4细胞+通过静脉过继转移Trp1 T细胞,并用106经照射的GVAX细胞(在其左侧腹腔注射)加200µgα-CTLA-4(腹腔注射)。在第6天和第8天,小鼠接受额外的GVAX和100µgα-CTLA-4治疗。在第10天通过CO处死小鼠2安乐死。为了进行保护实验,用10只4B16-BL6,并如上所述用GVAX和α-CTLA-4处理。每2-3天测量一次肿瘤。
组织处理和流式细胞术。
第10天用一氧化碳处死小鼠2将淋巴结(腹股沟、腋窝和肱骨)和肿瘤解剖成RPMI。淋巴结通过70µm过滤器分散,而肿瘤通过剪刀进行机械破坏,用无血清RPMI中0.33 mg/ml DNase(Sigma-Aldrich)和0.27 mg/ml Liberase TL(Roche)的混合物消化25分钟,然后通过70μm过滤器分散。为了鉴定T细胞群,用α-CD4 APC-eFluor780、α-CD45.1 eFluor 410、α-CD.45.2 APC、α-CTLA-4 PE和α-CD8 PerCP-eFluor710对样本进行染色。在一些实验中,使用了α-CD8明亮紫570和固定的活/死660染料。用Foxp3缓冲试剂盒(eBioscience)固定和渗透细胞,并用α-CD3 PE-Cy7、α-Foxp3 Alexa Fluor 700、α-CD3Pe-Cy7和α-Ki67 Alexa Fluor 488染色。在一些实验中,使用FASER试剂盒(Miltenyi Biotec)增强CTLA-4表面信号。为了鉴定巨噬细胞,样品用α-CD3 PerCP-eFluor710、α-Ly6C APC、α-Li6G PE-Cy7、α-NK1.1 APC、γ-CD11c PE、α-I-A Alexa Fluor700、α-CD45.2 eFluor410、α-FcγRIV Alexa488染色。样品与太平洋蓝微珠(生命科技公司)混合,以使样品体积正常化。数据通过LSR II和Fortessa流式细胞仪(BD)获得。每个肿瘤样本的绝对细胞数使用荧光微球进行量化,以归一化到已知的样本体积,然后将其归一化为肿瘤湿重。此计算使用以下公式:绝对细胞数=获得的细胞数/(肿瘤湿重/[获得的珠子数/添加到样品中的珠子数量])。
免疫荧光。
将肿瘤和淋巴结解剖成4%多聚甲醛,隔夜固定。样品在70%乙醇中脱水,包埋在石蜡中,并切成4µm的切片。切片用多克隆兔α-Iba1和多克隆大鼠α-Foxp3染色,然后用α-兔Alexa Fluor 488和α-大鼠Alexa Fluor 532进行二次染色。
SPR。
所有SPR分析均使用Biacore T100 SPR系统(GE Healthcare)在25°C的HBS EP+缓冲液(10 mM Hepes,pH 7.4,150 mM NaCl,3.4 mM EDTA,0.005%表面活性剂P20)中进行。在pH 4.5下,通过胺偶联将His标记的可溶小鼠FcγR胞外结构域(Sino Biological Inc.)固定在CM5芯片上,得到的密度为~2000 RU。将两倍连续稀释的9H10样品以30 ml/min的流速通过流式细胞注射3分钟,以进行结合,然后进行5分钟的分离阶段。对于FcγRIIB和FcγRIII结合分析,9H10样品的浓度范围为4000 nM至15.63 nM,对于Fc伽玛RI和Fc-γRIV结合分析,浓度范围为1000 nM至1.95 nM。在每个分析周期后,以50 ml/min的流速注入30 s优化浓度的NaOH,使传感器表面再生。减去与空白固定化流动细胞的背景结合和亲和常数Kd日使用BIAcore T100评估软件(1.1版)中建立的1:1结合动力学模型计算值。
统计分析。
使用Prism 5.0(GraphPad软件)分析数据。实验重复两到三次。统计显著性由Student’st吨测试(两组或两个条件之间)或ANOVA与事后测试(三个或更多组或条件)。采用Kaplan-Meier方法分析肿瘤生存数据。采用log-rank检验比较单变量分析中不同亚组的生存曲线。P值<0.05被认为具有统计学意义。
致谢
我们要感谢Tsvetelina Pencheva-Hoang和Andrew Furness对这份手稿的批判性审查。
T.R.Simpson获得了加拿大卫生研究院博士研究奖的支持。S.A.Quezada由英国癌症研究职业发展奖学金资助,是癌症研究所研究员奖的获得者。K.S.Peggs和F.Arce获得了英国白血病淋巴瘤研究所的资助。
J.P.Allison是加州大学授予Bristol Meyers Squib知识产权许可的发明人,也是Bristol Meyers Squibb的顾问。
作者没有进一步的财务利益冲突。
脚注
使用的缩写:
- ADCC公司
抗体依赖性细胞介导的细胞毒性
- CTLA-4型
细胞毒性T淋巴细胞相关抗原4
- GVAX公司
分泌粒细胞巨噬细胞集落刺激因子的肿瘤细胞疫苗
- SPR公司
表面等离子体共振
- T eff电池
CD4细胞+Foxp3系列−效应T细胞
- T调节细胞
CD4细胞+Foxp3系列+调节性T细胞
工具书类
-
Antony P.A.、Piccirillo C.A.、Akpinarli A.、Finkelstein S.E.、Speiss P.J.、Surman D.R.、Palmer D.C.、Chan C.-C.、Klebanoff C.A.、Overwijk W.W.等人。2005CD8+T细胞对肿瘤/自身抗原的免疫被CD4+T辅助细胞增强,并被天然存在的T调节细胞阻碍。免疫学杂志。174:2591–2601[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Benigni A.、Tomasoni S.、Turka L.A.、Longaretti L.、Zentilin L.、Mister M.、Pezzotta A.、Azzollini N.、Noris M.、Conti S.等人。2006腺相关病毒介导的CTLA4Ig基因转移可保护MHC不匹配肾移植免受慢性排斥反应。《美国肾脏学会杂志》。17:1665–167210.1681/ASN.2006010090[内政部] [公共医学] [谷歌学者]
-
陈A.、刘S.、Park D.、Kang Y.、Zheng G。2007通过FasL蛋白转移耗尽肿瘤内CD4+CD25+调节性T细胞可提高过继性T细胞转移的疗效。癌症研究。67:1291–129810.1158/0008-5472.CAN-06-2622[内政部] [公共医学] [谷歌学者]
-
Chen H.、Liakou C.I.、Kamat A.、Pettaway C.、Ward J.F.、Tang D.N.、Sun J.、Jungbluth A.A.、Troncoso P.、Logothetis C.、Sharma P。2009抗CTLA-4治疗导致非恶性和恶性前列腺组织中CD4+ICOShi T细胞频率和IFN-γ水平升高。程序。国家。阿卡德。科学。美国。106:2729–273410.1073/pnas.0813175106[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Clynes R.、Takechi Y.、Moroi Y.和Houghton A.、Ravetch J.V。1998Fc受体是黑色素瘤被动和主动免疫所必需的。程序。国家。阿卡德。科学。美国。95:652–65610.1073/第95.2652页[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Coe D.、Begom S.、Addey C.、White M.、Dyson J.、Chai J.G。2010. 抗GITR单克隆抗体耗尽调节性T细胞是癌症免疫治疗的一种新机制。癌症免疫学。免疫疗法。59:1367–137710.1007/s00262-010-0866-5[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Curran M.A.、Allison J.P。2009表达flt3配体的肿瘤疫苗与ctla-4阻断剂协同抑制植入前肿瘤。癌症研究。69:7747–775510.1158/0008-5472.CAN-08-3289[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
丰特诺J.D.、加文M.A.、鲁登斯基A.Y。2003Foxp3对CD4+CD25+调节性T细胞的发育和功能进行编程。自然免疫学。4:330–33610.1038/ni904[内政部] [公共医学] [谷歌学者]
-
Fossati-Jimack L.、Ioan-Facsinay A.、Reininger L.、Chicheportiche Y.、Watanabe N.、Saito T.、Hofhuis F.M.、Gessner J.E.、Schiller C.、Schmidt R.E.等人。2000抗红细胞自身抗体的四种免疫球蛋白G等型开关变体具有显著不同的致病性,这是基于它们在体内与低亲和力Fcγ受体III相互作用的能力。实验医学杂志。191:1293–130210.1084/jem.191.8.1293[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Giorgini A.、Brown H.J.、Lock H.R.、Nimmerjahn F.、Ravetch J.V.、Verbeek J.S.、Sacks S.H.、Robson M.G。2008Fc-γRIII和Fc-伽玛RIV对于开关变异单克隆抗体诱导的急性肾小球炎症是必不可少的。免疫学杂志。181:8745–8752[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Graeber M.B.、López-Redondo F.、Ikoma E.、Ishikawa M.、Imai Y.、Nakajima K.、Kreutzberg G.W.、Kohsaka S。1998新生大鼠面神经核轴切断后的小胶质细胞/巨噬细胞反应。大脑研究。813:241–25310.1016/S0006-8993(98)00859-2[内政部] [公共医学] [谷歌学者]
-
Griffiths R.W.、Elkord E.、Gilham D.E.、Ramani V.、Clarke N.、Stern P.L.、Hawkins R.E。2007肾癌患者调节性T细胞的频率及其与生存的相关性研究。癌症免疫学。免疫疗法。56:1743–175310.1007/s00262-007-0318-z[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
滨口Y.、秀Y.、高村K.、尼默尔贾恩F.、特德T.F。2006在CD20免疫治疗过程中,Fcγ受体的抗体等型特异性参与调节B淋巴细胞耗竭。实验医学学报203:743–7532005年10月10日[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Hirschorn-Cymerman D.、Budhu S.、Kitano S.、Liu C.、Zhao F.、Zhong H.、Lesokhin A.M.、Avogadri-Connors F.、Yuan J.、Li Y.等。2012CD4的肿瘤杀伤功能诱导+T细胞与伴随记忆和终末分化表型相关。实验医学学报209:2113–21262014年10月10日/20120532[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Hodi F.S.、Butler M.、Oble D.A.、Seiden M.V.、Haluska F.G.、Kruse A.、Macrae S.、Nelson M.、Canning C.、Lowy I.等人。2008抗体阻断细胞毒性T淋巴细胞相关抗原4对先前接种过疫苗的癌症患者的免疫学和临床效果。程序。国家。阿卡德。科学。美国。105:3005–301010.1073/pnas.0712237105[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Hodi F.S.、O'Day S.J.、McDermott D.F.、Weber R.W.、Sosman J.A.、Haanen J.B.、Gonzalez R.、Robert C.、Schadendorf D.、Hassel J.C.等人。2010转移性黑色素瘤患者使用伊普利单抗的生存率提高。北英格兰。医学杂志。363:711–72310.1056/NEJMoa1003466[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Kanda Y.、Yamada T.、Mori K.、Okazaki A.、Inoue M.、Kitajima-Miyama K.、Kuni-Kamochi R.、Nakano R.、Yano K.、Kakita S.等人。2007用三种不同的N-连接Fc寡糖(高甘露糖、杂交和复合型)比较非糖基化治疗性IgG1抗体的生物活性。糖生物学。17:104–11810.1093/乙二醇/cwl057[内政部] [公共医学] [谷歌学者]
-
Kavanagh B.、O'Brien S.、Lee D.、Hou Y.、Weinberg V.、Rini B.、Allison J.P.、Small E.J.、Fong L。2008CTLA4阻断以剂量依赖的方式扩大FoxP3+调节和激活效应CD4+T细胞。鲜血。112:1175–118310.1182/血液-2007-11-125435[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Kim J.M.、Rasmussen J.P.、Rudensky A.Y。2007调节性T细胞可在小鼠的整个生命周期内防止灾难性的自身免疫。自然免疫学。8:191–19710.1038/ni1428[内政部] [公共医学] [谷歌学者]
-
Klages K.、Mayer C.T.、Lahl K.、Loddenkemper C.、Teng M.W.L.、Ngiow S.F.、Smyth M.J.、Hamann A.、Huehn J.、Sparwasser T。2010. 选择性去除Foxp3+调节性T细胞可提高针对已确诊黑色素瘤的有效治疗性疫苗接种。癌症研究。70:7788–779910.1158/0008-5472.CAN-10-1736[内政部] [公共医学] [谷歌学者]
-
Krummel M.F.、Allison J.P。1996CTLA-4参与抑制IL-2积累和静止T细胞激活后的细胞周期进展。实验医学学报183:2533–254010.1084/jem.183.6.2533[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Leach D.R.、Krummel M.F.、Allison J.P。1996通过CTLA-4阻断增强抗肿瘤免疫。科学。271:1734–173610.1126/科学271.5256.1734[内政部] [公共医学] [谷歌学者]
-
李毅、叶聪。2008IL-21介导的Foxp3抑制导致抗原特异性CD8+细胞毒性T淋巴细胞生成增强。鲜血。111:229–23510.1182/血液-2007-05-089375[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Liakou C.I.、Kamat A.、Tang D.N.、Chen H.、Sun J.、Troncoso P.、Logothetis C.、Sharma P。2008CTLA-4阻断增加IFNgamma产生的CD4+ICOShi细胞,从而改变癌症患者中效应细胞与调节性T细胞的比率。程序。国家。阿卡德。科学。美国。105:14987–1499210.1073/pnas.0806075105[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Moo-Young T.A.、Larson J.W.、Belt B.A.、Tan M.C.、Hawkins W.G.、Eberlein T.J.、Goedegebuure P.S.、Linehan D.C。2009肿瘤衍生TGF-β介导小鼠胰腺癌模型中CD4+Foxp3+调节性T细胞的转化。免疫学杂志。32:12–2110.1097/CJI.0b013e318189f13c[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Muranski P.、Boni A.、Antony P.A.、Cassard L.、Irvine K.R.、Kaiser A.、Paulos C.M.、Palmer D.C.、Touloukian C.E.、Ptak K.等人。2008肿瘤特异性Th17-极化细胞可消除大型黑色素瘤。鲜血。112:362–37310.1182/血液-2007-11-120998[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Nimmerjahn F.、Ravetch J.V。2008Fcgamma受体作为免疫反应的调节器。国家免疫学修订版。8:34–4710.1038/nri2206[内政部] [公共医学] [谷歌学者]
-
Nimmerjahn F.、Bruhns P.、Horiuchi K.、Ravetch J.V。2005FcgammaRIV:一种具有独特IgG亚类特异性的新型FcR。免疫。23:41–5110.1016/j.immuni.2005.05.010[内政部] [公共医学] [谷歌学者]
-
Nimmerjahn F.、Lux A.、Albert H.、Woigk M.、Lehman C.、Dudziak D.、Smith P.、Ravetch J.V。2010FcγRIV缺失揭示了其在体内对IgG2a和IgG2b活性的中心作用。程序。国家。阿卡德。科学。美国。107:19396–1940110.1073/第101451107页[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Oflazoglu E.、Stone I.J.、Gordon K.A.、Grewal I.S.、van Rooijen N.、Law C.-L.、Gerber H.-P。2007巨噬细胞有助于抗CD30抗体SGN-30的抗肿瘤活性。鲜血。110:4370–437210.1182/血液-2007-06-097014[内政部] [公共医学] [谷歌学者]
-
Otten M.A.、van der Bij G.J.、Verbeek S.J.、Nimmerjahn F.、Ravetch J.V.、Beelen R.H.J.、van de Winkel J.G.J.和van Egmond M。2008肝转移瘤的实验性抗体治疗揭示了Fc-gammaRI和Fc-gammaRIV之间的功能冗余。免疫学杂志。181:6829–6836[内政部] [公共医学] [谷歌学者]
-
Peggs K.S.、Quezada S.A.、Chambers C.A.、Korman A.J.、Allison J.P。2009CTLA-4对效应和调节T细胞室的阻断有助于抗-CTLA-4抗体的抗肿瘤活性。实验医学学报206:1717–172510.1084/jem.20082492[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Piconese S.、Valzasina B.、Colombo M.P。2008OX40触发阻断调节性T细胞的抑制并促进肿瘤排斥反应。实验医学学报205:825–83910.1084/jem.20071341[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Quezada S.A.、Peggs K.S.、Curran M.A.、Allison J.P。2006CTLA4阻断和GM-CSF联合免疫治疗改变效应和调节性T细胞的瘤内平衡。临床杂志。投资。116:1935–194510.1172/JCI27745号[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Quezada S.A.、Peggs K.S.、Simpson T.R.、Shen Y.、Littman D.R.、Allison J.P。2008激活的T效应细胞对肿瘤的有限浸润限制了调节性T细胞耗竭对已建立的黑色素瘤的治疗活性。实验医学学报205:2125–213810.1084/jem.20080099[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Quezada S.A.、Simpson T.R.、Peggs K.S.、Merghoub T.、Vider J.、Fan X.、Blasberg R.、Yagita H.、Muranski P.、Antony P.A.等人。2010肿瘤反应CD4+T细胞在转移到淋巴减少宿主后产生细胞毒活性并消除已形成的大型黑色素瘤。实验医学学报207:637–65020091918年1月10日[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Quezada S.A.、Peggs K.S.、Simpson T.R.、Allison J.P。2011改变癌症免疫编辑的平衡:从肿瘤耐受到根除。免疫学。版次。241:104–11810.1111/j.1600-065X.2011.01007.x[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
阅读S.、Malmström V.、Powrie F。2000细胞毒性T淋巴细胞相关抗原4在CD25的功能中起重要作用+CD4细胞+控制肠道炎症的调节细胞。实验医学学报192:295–30210.1084/jem.192.2.295[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Read S.、Greenwald R.、Izcue A.、Robinson N.、Mandelbrot D.、Francisco L.、Sharpe A.H.、Powrie F。2006阻断CTLA-4对CD4+CD25+调节性T细胞的作用会使其体内功能丧失。免疫学杂志。177:4376–4383[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
参考M.E.、Carner K.、Chambers K.S.、Chinn P.C.、Leonard J.E.、Raab R.、Newman R.A.、Hanna N.、Anderson D.R。1994CD20嵌合鼠-人单克隆抗体体内B细胞耗竭。鲜血。83:435–445[公共医学] [谷歌学者]
-
Robert C.、Thomas L.、Bondarenko I.、O'Day S.、M D J.W.、Garbe C.、Lebbe C.、Baurain J.F.、Testori A.、Grob J.J.等人。2011依匹单抗联合达卡巴嗪治疗未经治疗的转移性黑色素瘤。北英格兰。医学杂志。364:2517–252610.1056/NEJMoa1104621[内政部] [公共医学] [谷歌学者]
-
Schmidt E.M.、Wang C.J.、Ryan G.A.、Clough L.E.、Qureshi O.S.、Goodall M.、Abbas A.K.、Sharpe A.H.、Sansom D.M.、Walker L.S.K。2009Ctla-4控制调节性T细胞外周稳态,是抑制胰岛自身免疫所必需的。免疫学杂志。182:274–282[内政部] [公共医学] [谷歌学者]
-
Selby M.J.、Engelhardt J.J.、Quigley M.、Henning K.A.、Chen T.、Srinivasan M.、Korman A.J。2013IgG2a同型抗CTLA-4抗体通过减少肿瘤内调节性T细胞增强抗肿瘤活性。癌症免疫学研究。10.1158/2326-6066.CIR-13-0013[内政部] [公共医学] [谷歌学者]
-
Setiady Y.Y.、Coccia J.A.、Park P.U。2010PC61抗CD25单克隆抗体对CD4+FOXP3+Treg细胞的体内耗竭由FcgammaRIII+吞噬细胞介导。欧洲免疫学杂志。40:780–78610.1002/eji.200939613[内政部] [公共医学] [谷歌学者]
-
Shi C.、Hohl T.M.、Leiner I.、Equinda M.J.、Fan X.、Pamer E.G。2011. Ly6G+中性粒细胞对于防御系统性单核细胞增生性李斯特菌感染是必不可少的。免疫学杂志。187:5293–529810.4049/jimmunol.1101721[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Shrikant P.、Khoruts A.、Mescher M.F。1999. CTLA-4阻断通过CD4+T细胞和IL-2依赖机制逆转CD8+T细胞对肿瘤的耐受性。免疫。11:483–49310.1016/S1074-7613(00)80123-5[内政部] [公共医学] [谷歌学者]
-
Stockmeyer B.、Elsässer D.、Dechant M.、Repp R.、Gramatzki M.、Glennie M.J.、van de Winkel J.G.、Valerius T。2001Fc受体导向的双特异性抗体对G-CSF或GM-CSF刺激肿瘤细胞杀伤的机制。免疫学杂志。方法。248:103–11110.1016/S0022-1759(00)00346-X[内政部] [公共医学] [谷歌学者]
-
Stuart L.M.、Ezekowitz R.A.B。2005吞噬作用:优雅的复杂性。免疫。22:539–55010.1016/j.immuni.2005.05.002[内政部] [公共医学] [谷歌学者]
-
Sutmuler R.P.、van Duivenvoorde L.M.、van-Elsas A.、Schumacher T.N.、Wildenberg M.E.、Allison J.P.、Toes R.E.、Offringa R.、Melief C.J。2001细胞毒性T淋巴细胞相关抗原4阻断和CD25(+)调节性T细胞在抗肿瘤治疗中的耗竭的协同作用揭示了抑制自身反应性细胞毒性T细胞反应的替代途径。实验医学学报194:823–83210.1084/jem.194.6.823[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
斯旺森J.A.,霍普A.D。2004Fc受体介导吞噬过程中的信号协调。J.Leukoc。生物。76:1093–110310.1189/jlb.0804439[内政部] [公共医学] [谷歌学者]
-
Takahashi T.、Tagami T.,Yamazaki S.、Uede T.、Shimizu J.、Sakaguchi N.、Mak T.W.、Sakaughi S。2000组成性表达细胞毒性T淋巴细胞相关抗原4的CD25(+)CD4(+)调节性T细胞维持免疫耐受性。实验医学学报192:303–31010.1084/jem.192.2.303[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Takai T.、Li M.、Sylvestre D.、Clynes R.、Ravetch J.V。1994FcRγ链缺失导致多营养效应细胞缺陷。单元格。76:519–52910.1016/0092-8674(94)90115-5[内政部] [公共医学] [谷歌学者]
-
特德·T·F、巴拉斯·A、秀·Y。2006. Fcgamma受体依赖性效应器机制调节CD19和CD20抗体免疫治疗B淋巴细胞恶性肿瘤和自身免疫。施普林格·塞米恩。免疫病理学。28:351–3642007年10月1日/00281-006-0057-9[内政部] [公共医学] [谷歌学者]
-
Uchida J.、Hamaguchi Y.、Oliver J.A.、Ravetch J.V.、Poe J.C.、Haas K.M.、Tedder T.F。2004在抗CD20抗体免疫治疗期间,先天性单核吞噬细胞网络通过Fc受体依赖机制耗尽B淋巴细胞。实验医学学报199:1659–166910.1084/jem.20040119[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
van Elsas A.、Hurwitz A.A.、Allison J.P。1999使用抗细胞毒性T淋巴细胞相关抗原4(CTLA-4)和产生粒细胞/巨噬细胞集落刺激因子(GM-CSF)的疫苗对B16黑色素瘤进行联合免疫治疗,可诱导皮下和转移性肿瘤的排斥反应,并伴有自身免疫性脱色。实验医学学报190:355–36610.1084/jem.190.3.355[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Waitz R.、Solomon S.B.、Petre E.N.、Trumble A.E.、FassóM.、Norton L.、Allison J.P。2012联合肿瘤冷冻消融和抗CTLA-4治疗有效诱导肿瘤免疫。癌症研究。72:430–43910.1158/0008-5472.CAN-11-1782[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Wang S.-Y.、Weiner G。2008肿瘤抗体治疗中的补体和细胞毒性。专家操作。生物疗法。8:759–76810.1517/14712598.8.6.759[内政部] [公共医学] [谷歌学者]
-
Wing K.、Onishi Y.、Prieto-Martin P.、Yamaguchi T.、Miyara M.、Fehervari Z.、Nomura T.、Sakaguchi S。2008CTLA-4对Foxp3+调节性T细胞功能的控制。科学。322:271–27510.1126/科学.1160062[内政部] [公共医学] [谷歌学者]
-
Xie Y.、Akpinarli A.、Maris C.、Hipkiss E.L.、Lane M.、Kwon E.-K.M.、Muranski P.、Restifo N.P.、Antony P.A。2010体内分化的天然肿瘤特异性CD4(+)T细胞可消除已建立的黑色素瘤。实验医学学报207:651–6672009年9月10日至1921年1月10日[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Zhou X.、Bailey-Bucktrout S.L.、Jeker L.T.、Penaranda C.、Martínez-Lolrdella M.、Ashby M.、Nakayama M.、Rosenthal W.、Bluestone J.A。2009转录因子Foxp3的不稳定性导致体内产生致病性记忆T细胞。自然免疫学。10:1000–100710.1038/ni.1774年10月10日[内政部] [PMC免费文章] [公共医学] [谷歌学者]

