摘要 与阿尔茨海默病(AD)相关的特征性神经病理学变化和其他证据支持淀粉样蛋白级联假说。 将淀粉样蛋白沉积视为痴呆症的主要诱因,现在已经导致了多种策略的临床试验,以消除或防止其形成。 我们对3名接受巴匹诺单抗输注治疗的受试者进行了神经病理学和生物化学评估。 组织学分析用于量化淀粉样斑块密度、Braak分期和大脑淀粉样血管病(CAA)的程度。 用ELISA法对额叶和颞叶标本中淀粉样β(Aβ)物种进行定量。 对淀粉样蛋白-β前体蛋白(AβPP)及其C末端(CT)片段和τ物种进行了蛋白质印迹。 将Bapineuzumab治疗的受试者(Bapi-AD)与非免疫年龄匹配的AD(NI-AD)和非痴呆对照(NDC)受试者进行比较。 我们的研究表明,Bapi-AD患者的总淀粉样斑块密度与NI-AD患者相似。 此外,NI-AD和Bapi-AD患者的CAA为中度至重度。 尽管组织学上可显示的软脑膜、脑血管和神经实质-淀粉样体密度均未受治疗影响,但Bapi-AD受试者的Aβ肽谱发生显著改变。 总aβ有减少的趋势 42 以及Aβ的增加 40 导致aβ相应显著下降 42 :Aβ 40 与NI-AD受试者的比率。 Bapi-AD和NI-AD受试者之间的AβPP、CT99和CT83或tau物种水平没有差异。 Aβ谱的显著变化揭示了淀粉样蛋白的动态生成,其中的去除和沉积过程明显受到巴匹诺单抗治疗的干扰。 尽管生化成分发生了变化,但所有3名免疫受试者的认知能力都持续下降。
介绍 痴呆症最常见的形式是阿尔茨海默病,目前全球约有2400万人患有阿尔茨海默病,预计发病率每20年翻一番 [1] 该病的神经病理学特征是纤维淀粉样β(Aβ)肽、淀粉样斑块(AP)和脑淀粉样血管病(CAA)的大量沉积,以及主要由tau蛋白组成的神经纤维缠结(NFT)的神经元内积聚。 这些病变的丰富性支持了淀粉样级联假说,认为淀粉样级联是AD发病的根本原因。由于早老蛋白(PS)和淀粉样β前体蛋白(AβPP),家族性AD(FAD)中也存在AP、CAA和NFT,这一模型得到了加强 突变和在携带AβPP、PS和tau突变形式的基因工程转基因(Tg)小鼠中重演。 此外,携带3份21号染色体(AβPP基因的位置)的唐氏综合征患者也会发展为AD的神经病理学。
目前,还没有有效的AD疾病修饰治疗方法。然而,不溶性Aβ的大量沉积和可溶性低聚物Aβ的升高促使设计了多种主动和被动免疫疗法,以消除这些肽的毒性形式 [2] , [3] 被动免疫治疗的一个例子是bapineuzumab,一种人源化单克隆抗体(3D6),专门针对aβ的N末端区域(残基1-5) [4] 尽管被动和主动免疫疗法在AβPP和PS Tg小鼠模型中对Aβ的去除是有效的,但从神经病理学的角度来看,这些干预措施在AD患者中的应用仅部分成功 [5] – [9] 虽然在临床试验中没有观察到对疾病过程有明显的有益改变 [7] , [10] , [11] .
在这项研究中,我们评估了3名参与评估巴普奈珠单抗临床试验的患者的临床病史、神经病理学和生化结果 [12] (临床试验.gov标识符 NCT00112073号 )接受2、6或20次免疫治疗剂量的患者。 我们量化并表征了巴匹诺单抗免疫性AD(Bapi-AD)患者额叶和颞叶中残留的可溶性和不溶性Aβ肽的水平,并将其与4名年龄匹配的非免疫性AD和4名年龄配对的非痴呆对照(NDC)个体进行了比较。 还定量了AβPP及其C末端(CT)肽和细胞因子肿瘤坏死因子-α(TNF-α)。 我们还将bapineuzumab的观察结果与接受AN-1792免疫治疗的受试者的观察结果进行了比较。
材料和方法 临床和神经病理报告及治疗简介 本报告涉及对3名Bapi-AD患者进行的神经病理学和生物化学观察,以及与NI-AD和NDC患者的比较。 病例#1和#2的大脑样本来自Banner Sun Health Research Institute(BSHRI)的大脑和身体捐赠计划 [13] 其业务已获得Banner Health Institutional Review Board的批准。 所有参加大脑和身体捐赠计划的受试者签署一份由Banner Health Institutional Review Board批准的知情同意书。 病例3的脑组织由记忆障碍和神经疾病研究所和加州大学阿尔茨海默病研究中心(UCI-ADRC)提供。 参加UCI-ADRC的参与者提供经UCI机构审查委员会批准的知情同意书。 主题人口统计学和神经病理学摘要见
表1 .
表1。 受试者人口统计学和神经病理学评估。
国家数据中心 过期年龄(年) 性别 PMI(小时) 脑重(g) 上次MMSE得分
APOE公司 燃气轮机
病程(年) 斑块总得分 菌斑密度 制动阶段 CAA公司 WMR总分
20 78 F类 3.33 1150 28 2/3 – 4 稀疏的 三 轻度 2
21 90 F类 4 1160 26 3/3 – 0 零 四、 轻度 0
22 76 M(M) 2.33 1375 29 3/4 – 5.5 稀疏的 我 轻度 1
23 74 M(M) 3.25 1440 不适用 2/3 – 0 零 我 无 2
NI-AD公司
过期年龄(年)
性别
采购经理人指数(小时)
大脑重量 (g)
上次MMSE得分
APOE公司 燃气轮机
病程(年)
斑块总得分
菌斑密度
制动阶段
CAA公司
WMR总分
10 91 F类 2 1045 不适用 3/4 7 14 频繁的 V(V) 轻度 不适用
11 84 M(M) 2 1160 16 4/4 8 14 频繁的 V(V) 无 0
12 87 M(M) 2.5 1100 13 2/3 6 15 频繁的 不及物动词 重度Occ。 6
13 103 F类 2.6 930 4 2/3 10 13.5 频繁的 四、 重度Occ。 7
BSHRI Bapi-AD公司
过期年龄(年)
性别
PMI(小时)
大脑重量 (g)
上次MMSE得分
APOE公司 燃气轮机
病程(年)
斑块总得分
菌斑密度
制动阶段
CAA公司
WMR总分
1 79 F类 三 1000 9 4/4 8 15 频繁的 不及物动词 严重阻塞/ 平价。 5
2 89 M(M) 2.2 1132 21 2/3 11 12.5 频繁的 V(V) 国防部。 2
UCI Bapi-AD公司
过期年龄(年)
性别
采购经理人指数(小时)
脑重(g)
上次MMSE得分
APOE公司 燃气轮机
病程(年)
斑块总得分
菌斑密度
制动阶段
CAA公司
WMR总分
三 86 M(M) 5 1170 0 3/4 8 不适用 频繁的 不及物动词 国防部。 Occ.公司/ 正面 不适用
案例#1 有关病例#1的详细临床病理描述,请参阅我们最近的出版物 [14] 简言之,一名79岁的女性在死亡前8年左右被诊断为AD。 在临床试验的延长期内,患者在39周内接受了4剂bapineuzumab(每剂0.5 mg/kg)。 在最后一次服药后约1个月,患者在MRI扫描中显示出血管源性水肿的症状和体征,在她去世之前,MRI扫描已被清除。 没有明显的疾病改变迹象可以归因于免疫治疗。 神经病理学分析显示频繁发生AP和NFT,同时诊断为AD和Binswanger型血管性痴呆。 病例#1被纳入病例#2和#3的当前生化评估中,以增加样本量。 此外,在之前对案例#1的分析中,我们只描述了额叶区域,而在这次交流中,我们添加了时间区域。
案例2:临床病史 一名89岁男子在症状出现约12年后死于AD。 现有的私人医疗记录包括他在去世前11年左右首次出现的情况,当时一名神经科医生对他进行了检查,发现他在颞动脉炎活检后有几个月的认知功能恶化史。 即使在离家很近的地方散步,他也会迷路,并且经常重复自己的话。 此外,他正在接受抑郁症治疗。 考试时他不适应这个位置。 他在简易精神状态检查(MMSE)中得了23/30分,在延迟回忆和定向方面失分。 步态和姿势正常,无局部神经症状。 脑MRI仅显示左椎动脉颅内段扩张。 EEG被解释为在正常范围内。 他开始服用Aricept,用于早期阿尔茨海默氏痴呆症的推定诊断,但由于胃肠道副作用,很快就改用Exelon。 在接下来的10年里,他的认知状态下降非常缓慢,MMSE得分为20/30、19/30,死亡前大约1个月的最终得分为21/30。 在这段时间里,他出现了攻击性行为,并接受了抗精神病药物治疗。 他出现了惊吓性肌阵挛、弯腰姿势和颤抖。 脑电图只显示出缓慢和紊乱。 大约在死亡前1年,他有直立性低血压伴晕厥发作,死亡前几个月因心动过缓住院。 2003年至2011年,他在BSHRI接受了年度标准化神经和神经心理学评估。 诊断印象是可能的AD引起的痴呆,伴有步态共济失调和原发性震颤。 他的既往病史以高脂血症、二尖瓣脱垂、一度房室传导阻滞、慢性阻塞性肺病、糖耐量异常、甲状腺结节、双侧白内障摘除术、青光眼和良性前列腺肥大而著称。 他的家族史以他母亲的晚发性痴呆症而闻名。
2006年1月至2007年1月,该患者被纳入了bapineuzumab临床试验AAB-201(临床试验。政府标识符 NCT00112073号 ). 2007年4月,该患者开始参与bapineuzumab开放标签临床试验AAB-001,并在该项目中一直持续到2011年1月。 在此期间,该患者在260周内接受了20次bapineuzumab输注(每次1 mg/kg)。 每次输注bapineuzumab大约每3个月进行一次。 载脂蛋白E( APOE公司 )该个体的基因型为ε2/3。
2003年、2004年、2009年和2011年的神经心理学数据可用。 在记忆领域,Rey AVLT总学习从低平均水平持续下降至轻度受损。 在任何测试年份的延迟中,他都无法回忆起任何AVLT信息。 识别记忆原始分数有所不同,但多年来都在受损范围内。 叙事回忆(WMS-R逻辑记忆)于2009年和2011年进行,两年中仅发现轻度至中度损伤。 视觉记忆(BVMT-R)仅在2009年和2011年使用,这两年的表现均为中度至重度受损。 在四个测试期内,简单视觉注意力(TMTA)从低平均水平下降到中度受损。 简单听觉注意(数字向前)从2003年和2004年的轻度受损改善到2009年和2011年的低平均水平; 这是跨度提高一点的差异。 执行功能(根据Stroop C/W和Trail Making Test B进行测量)随着时间的推移从低平均值下降到中度至重度损伤。 对峙命名从2009年的26/30个正确识别对象减少到2011年的21/30个。 在前3个测试年中,线路方向判断(JLO)处于平均范围。 由于无法完成样本项目,2011年未对JLO进行管理。 2011年,受试者开始出现颜色辨别问题。 MMSE(分别为24、21、24、21)和时钟绘制任务(每年10/10)表现一致。 语言流利度在不同时期是不同的。 2003年和2004年,语音流利度处于平均水平,高于2009年的平均水平,2011年处于平均范围的低端。 2003年,动物的语义流利性轻度受损,2004年平均水平较低,2009年平均水平,2011年平均水平偏低。 在8年的时间里,该科目的认知水平普遍呈下降趋势,但个别测试例外。
案例2:神经病理学报告 粗略检查 尸检时大脑重量为1132克。硬脑膜正常,软脑膜中度纤维化。 凸起是对称的,表现为额叶中度脑回萎缩,顶叶中度至重度脑回萎缩,枕叶无脑回萎缩。 大脑凸面或基底部无局灶性病变。 Willis循环显示中度斑块状动脉粥样硬化。 乳头体的形状、颜色和大小正常。 颞叶和unci表现为轻度至中度的脑回萎缩。 小脑和脑干外观正常。 脑切片显示侧脑室后角中度至明显增大,其他部位轻度扩张。 基底神经节、丘脑和丘脑底核不明显。 杏仁核左半球中度萎缩,右半球严重萎缩,颞角代偿性增大。 两个半球的海马体头部轻度萎缩。 海马体和海马旁回均轻度萎缩。 双侧黑质轻度脱色。 脑干和小脑的轴位和副矢状位切片均正常。
显微镜检查 用苏木精和伊红(H&E)染色的左侧大脑石蜡切片显示,在大脑皮层切片中,轻度至中度上层胶质增生。 杏仁核和内嗅皮层显示中度至明显的胶质增生。 海马CA1区轻度胶质增生。 基底神经节不明显。 下丘脑轻度至中度胶质增生。 包括丘脑底核和乳头体在内的丘脑底区域不明显。 黑质没有明显的色素神经元耗竭,而蓝斑中度至显著耗竭; 这两个地区都没有路易尸体。 小脑上蚓部出现中度至明显的Purkinje细胞斑片状丢失。 小脑、脑干和主要脊髓的剩余部分无明显变化。 H&E染色的大切片显示脑白质无明显稀疏,无梗死。 苍白球内有几条矿化血管。 Gallyas、Campbell-Switzer和Thioflavine-S方法染色的切片显示,在新皮质区,弥漫型老年斑块频繁出现,而神经炎和核心斑块呈斑块状分布,在额叶、顶叶和枕叶从稀疏到频繁不等,在颞叶中密度适中到频繁。 神经纤维缠结也呈片状分布,在新皮质区从稀疏到中度到局部频繁。 杏仁核、内嗅皮层和海马CA1区经常出现缠结。 杏仁核、内嗅皮质和海马CA1区中的嗜银颗粒密度较高。 杏仁核周围常有Gallyas阳性胶质细胞; 这些类似于具有尖峰突起的小星形胶质细胞。 大脑淀粉样血管病在大多数大脑皮层区域表现为稀疏到中度到局灶性密度,而小脑软脑膜则表现为局灶性中度密度。 磷酸化α-突触核蛋白的免疫组织化学染色显示,嗅球、脑干、杏仁核或大脑皮层中没有免疫反应性内含物或相关神经突起。 诊断:阿尔茨海默病; 嗜银颗粒和非特异性胶质tau病,颞叶内侧。 评论:这一显微镜检查证实了AD的临床诊断。嗜银颗粒是一个意义不明的显微镜发现; 大约25%的认知功能正常的老年人以及AD和其他老年脑疾病患者中的类似比例都会出现这种情况。 在这种情况下,他们通常伴有非特异性的神经胶质tau病变。
案例3:临床病史 该患者为86岁男性,接受过12年教育,在1999年UCI-ADRC首次评估之前,曾有4年突发记忆障碍史。 他被ADRC临床核心纵向跟踪,病情缓慢恶化。 该患者于2006年8月接受了bapineuzumab临床试验的筛查,并参加了这项具有中度认知障碍的双盲随机多中心研究(MMSE 17/30)。 该患者仅接受了2次剂量的2.0 mg/kg bapineuzumab。 第一次输液于2006年9月进行,13周后第二次输液。 每次输注后6周完成MRI扫描以进行安全性评估,第二次输注后6周在安全性扫描中观察到急性血管源性水肿。 死亡前6个月测得的MMSE为8/30,出现渐进性认知下降。 尸检时,观察到Braak VI期患者有足够的神经病理学诊断为患有路易体病的AD APOE公司 该个体的基因型为ε3/4。
案例3:神经病理学报告 粗略检查 尸检时大脑重量为1170克。外观检查时,大脑脑回和脑沟的大体外观正常。 大脑动脉及其位于大脑底部的主要分支显示中度局灶性动脉粥样硬化,管腔狭窄达40%。 右侧大脑冠状切片显示侧裂区存在轻微的脑回狭窄和脑沟增宽。 尽管海马体较小,但侧脑室似乎没有扩大。 脑干和小脑的水平切片显示,黑质内的神经黑色素沉着程度正常,蓝斑内的色素沉着强度显著降低。
显微镜检查 从额叶中部、颞叶上部、顶叶下部、跟骨/心包石、头侧和尾侧扣带回皮质和白质以及海马、杏仁核、纹状体、丘脑、中脑、脑桥、髓质和小脑检查组织块。 弥漫性和神经炎(Braak和Braak C期)斑块在额叶、颞叶、顶叶、枕叶、头侧和尾侧扣带回皮质以及海马CA1区、脑室下、鼻内-鼻腔区和杏仁核内形成强烈。 在大脑新皮层内,浅层内的神经炎斑块(在改良的Bielschowsky和Aβ染色切片中)比深层内更突出、染色更密集,而斑块的数量似乎相似,但染色强度较轻。 这些发现也在海马结构中观察到,尤其是在脑室下,更引人注目的是在杏仁核内,许多神经炎斑块内的Aβ免疫染色强度显著降低。 在额叶、枕叶软脑膜和皮质内血管中也观察到中度血管壁内Aβ沉积,在杏仁核毛细血管中也有轻度血管壁内沉积。 在Perl的铁污渍的帮助下,铁沉积是最小的。 神经纤维变性被认为是Braak VI期,在所有检查部位都严重,但跟骨皮质除外,那里没有神经纤维变性。 在海马CA1内很容易观察到颗粒卵巢变性和平野小体形成。 黑质内神经黑色素承载神经元的浓度正常,蓝斑内中度严重降低。 黑质、杏仁核、黑质下、内嗅皮质和吻侧扣带回皮质内均可见Alpha-synuclein免疫阳性的胞质内Lewy包涵体。
神经病理学评估 对Bapi-AD患者#1和#2、NI-AD和NDC受试者(BSHRI)进行斑块总积分、淀粉样斑块密度、CAA总积分、NFT总积分、白质稀疏(WMR)积分、CERAD标准、神经炎斑块积分的评估 [15] 和Braak舞台 [16] 如以前的出版物中详细描述的 [13] , [17] , [18] 作为标准UCI-ADRC神经病理学核心方案的一部分,将Bapi-AD病例#3的固定组织块包埋在石蜡中,并在8µm处切片,以进行最终标准神经病理学诊断(NIA-Reagan标准 [19] ). 使用标准的免疫组织化学方法应用改良的Bielschowsky、H&E和Klüver-Barrera染色和tau、α-突触核蛋白、泛素、胶质纤维酸性蛋白(GFAP-星形胶质细胞)和CD68(小胶质细胞)的免疫染色 [20] , [21] .使用银色切片评估Braak分期 [16] 为了进行额外的实验研究,用振捣器以50µm的间隔对包含额叶中回和海马的组织块进行切片。 采用标准免疫组织化学方法检测Aβ 1–40 和Aβ 1–42 (Biosource International,Camarillo,CA)用90%甲酸预处理4分钟后 [22] 使用硫黄烷-S(0.1%)染色观察含有Aβ的现有斑块是否为纤维状。
脑血管淀粉样血管病(CAA)的评估 为了研究软脑膜血管淀粉样变的程度,在脑冠状切片之前,从大脑半球的凸面和内侧小心地去除这些膜。 软脑膜在磷酸盐缓冲盐水(PBS)中冲洗,并在−80°C下冷冻,直至使用。 为了清除夹带的血管内血液,分别用2 L冷PBS冲洗软脑膜6次,最后用2 L去离子水(DW)冲洗。 将闪闪发光的膜铺在3个塑料培养皿(直径14cm)的表面上,并通过在65°C的烤箱中加热使其干燥并粘附在塑料表面。 用无水乙醇固定膜1 h,用DW冲洗,用1%水溶性硫黄烷-S染色15 min,然后用70%乙醇冲洗4次,以去除未结合的荧光色素,并立即在外荧光显微镜上观察。 用于评估皮质血管淀粉样蛋白,0.5 cm 三 将大脑皮层悬浮在含有5%十二烷基硫酸钠和0.01%叠氮化钠的600 ml 50 mM Tris-HCl pH 7.5缓冲液中,持续搅拌72 h,直到所有组织溶解,不溶性血管除外。 用4 L DW冲洗血管,以去除多余的SDS,并按照上述方法对软脑膜进行处理和染色。
免疫组织化学 使用滑动冷冻切片机从Bapi-AD病例#1和#2、NI-AD和NDC受试者中采集40µm厚的冠状游离福尔马林固定切片,并在PBS、0.3%Triton X-100(PBS-Tx)缓冲液中清洗,以去除冷冻保护剂存储介质。 对于二氨基联苯胺(DAB)信号显影,在1%H中阻断各节段30分钟 2 O(运行) 2 用PBS-Tx洗涤,并在室温下在各自由Abcam提供的一级抗体中孵育16 h:抗CD68(ab955;1∶1000稀释)、抗HLA-DR(ab2018;1∶000稀释)和抗CD3(ab16669;1∶500稀释)。 然后在PBS-Tx中清洗切片,在适当的次级生物素化抗体中培养2 h,用PBS-Tx清洗切片,并在亲和素-生物素过氧化物酶(ABC,Vector Labs)中培养30 min。 将切片放置在DAB中3-8分钟,转移到Tris-buffer中,并安装在玻璃载玻片上,然后用1%中性红进行复染并脱水。 这些部分用Permount安装介质覆盖(宾夕法尼亚州匹兹堡Fisher Scientific)。 为了进行荧光染色,将切片与兔抗GFAP(ab7260:Abcam,Cambridge,MA)以1∶3000稀释液反应,在室温下在摇床上培养20 h,然后用PBS-Tx洗涤6次。将切片与Alexa Fluor 488-共轭山羊抗兔IgG孵育2 h(a-11034:Life Technology Corp。 Carlsbad CA)以1∶1000稀释,然后在PBS-Tx中清洗6次,将切片安装在玻璃载玻片上并干燥。 然后将载玻片依次浸入70%乙醇、1%苏丹黑70%乙醇、50%乙醇中,用dH冲洗 2 O,并用Vectashield硬装介质(Vector Labs,Burlingame CA)安装。
ELISA法测定可溶性和不溶性Aβ 先前已发布了ELISA方法的详细说明 [23] 简而言之,将额叶和颞叶灰质(100 mg)在800µl 20 mM Tris-HCl、5 mM EDTA、pH 7.8、蛋白酶抑制剂混合物(PIC,罗氏诊断公司,德国曼海姆)中轻轻均质,在435000× 克 以及收集的Tris-HCl可溶性上清液。 在600µl 90%的玻璃蒸馏甲酸(GDFA)中重新配制颗粒,在435000× 克 收集上清液,用去离子水透析,然后用0.1 M碳酸氢铵去除GDFA,然后冻干。 在500µl 5 M盐酸胍(GHCl)、50 mM Tris-HCl、pH 8.0、PIC(Roche)中重新配制冻干材料,离心并收集上清液。 使用Pierce的Micro-BCA蛋白质检测试剂盒测定Tris和GDFA/GHCl可溶性提取物的总蛋白质。 Aβ 40 和Aβ 42 根据制造商说明,使用Invitrogen(加利福尼亚州卡尔斯巴德)的ELISA试剂盒进行定量。
ELISA法测定肿瘤坏死因子-α(TNF-α) 将灰质(100 mg)均匀化于10体积的20 mM HEPES、1.5 mM EDTA、pH 7.4、PIC(罗氏)中,如之前所述 [23] 和总蛋白浓度测定(BCA蛋白分析,Pierce)。 按照制造商的指示,使用ELISA试剂盒(德国海德堡PromoKine)对人TNF-α水平进行量化。
Western Blot分析 蛋白质印迹的完整材料和方法已在别处出版 [23] 用于这些实验的抗体是CT20AβPP(针对AβPP的最后20个氨基酸;新泽西州普林斯顿市科万斯市#SIG-39152)和tau(针对tau的159-163个氨基酸的HT7克隆;伊利诺伊州Pierce市Rockford市#MN1000)。 对于二级抗体,使用了HRP偶联的AffiniPure山羊抗鼠IgG(#111-035-144,宾夕法尼亚州西格罗夫Jackson ImmunoResearch)或HRP偶联AffiniPure山羊抗兔IgG。 此外,使用α-小鼠肌动蛋白(#A65020,BD转导实验室,加利福尼亚州圣何塞)或α-兔肌动蛋白(#Ab37063,Abcam,马萨诸塞州剑桥)作为总蛋白负载对照。
统计分析 使用GraphPad Prism 5软件(加利福尼亚州La Jolla)进行统计计算,使用单向方差分析,然后进行Tukey多重比较测试。 统计显著性由以下因素决定 第页 值≤0.05。
结果
表 1 显示了年龄、性别、尸检间隔、大脑重量、上次MMSE得分的详细说明, APOE公司 本研究中使用的NDC、NI-AD和Bapi-AD病例的基因型和神经病理学评估。 在NDC组和NI-AD组中,各有2名女性和2名男性,而Bapi-AD组有1名女性(病例#1)和2名雄性(病例#2和#3)。 这些组中个体的平均年龄分别为80岁、91岁和85岁。 Bapi-AD患者的平均脑重为1101克,与NI-AD患者平均体重(1059克)非常接近,与NDC组的平均体重(1281克)相比,提示有一定程度的萎缩。 Bapi-AD受试者的最终MMSE平均值为10,而NI-AD和NDC MMSE值分别为11和28。 这个 APOE公司 研究中的3组ε4等位基因频率分别为:NDC=13%、NI-AD=38%和Bapi-AD=50%。 NDC患者的AP密度为零至稀疏,而NI-AD和Bapi-AD患者的AP浓度较高 (
表1
) ,可以在
图1 和
图2 NDC组和NI-AD/Bapi-AD组淀粉样斑块升高(
表1 ). NI-AD和Bapi-AD患者的神经纤维缠结Braak分期相似(
表1 ). CAA含量差异很大 (
表1
) :NDC无至轻度,NI-AD无至轻度(#12和#13病例枕叶严重),Bapi-AD中至重度(#1病例枕叶和顶叶严重)。 如所示
表1 ,三组的WMR总分差异较大。
图1。 Bapi AD和NI-AD中的淀粉样斑块和血管淀粉样蛋白。
额叶淀粉样斑块( A类 , C类 和 E类 )和时间( B类 , D类 和 F类 )叶和血管淀粉样蛋白( G公司 和 H(H) ). A类 )和 B类 )Bapi-AD病例#2的Campell-Switzer染色(89岁男性, APOE公司 ε2/3基因型)。 C) 和 D) 12号NI-AD病例的Campbell-Switzer染色(87岁男性, APOE公司 ε2/3). E类 )和 F类 )Bapi-AD病例#2的硫黄烷-S染色。 所有病例均可见频繁的淀粉样斑块。 G公司 )Bapi-AD病例#2的脑膜用硫黄S染色。 H(H) )Bapi-AD病例#2的皮质血管用硫黄烷-S染色。 A–F 以100倍的放大倍数拍摄,比例尺如标题所示 A类 . G公司 和 H(H) 以25倍的倍数拍摄,每个标题中都显示了比例尺。
图2。 Bapi AD和NI-AD中的淀粉样斑块和血管淀粉样蛋白。
额叶淀粉样斑块( A、 C、E )和时间( B、 D、F )叶和血管淀粉样蛋白( G公司 和 H(H) ). A类 )和 B类 )Bapi-AD病例#3的Bielschowsky染色(86岁男性, APOE公司 ε3/4基因型),显示中度淀粉样斑块积聚。 C类 )和 D类 )NI-AD病例#10的Campbell-Switzer染色(91岁女性, APOE公司 ε3/4基因型),表现出频繁的淀粉样斑块。 E类 )Bapi-AD病例#3额叶皮质的硫黄烷-S染色显示淀粉样斑块和背景NFT。 F类 )抗Aβ抗体对Bapi-AD病例#3淀粉样斑块的双重免疫标记 40 (棕色)和Aβ 42 (蓝色)表明淀粉样斑块包含这两种肽。 Bapi-AD病例#3的皮质血管( G公司 )和Bapi-AD案例#1( H(H) )用硫黄烷-S染色。放大倍数: A、 C、D、E、F –100倍和 B类 –40倍。
如您所愿
图1A、B、E、F 和
图2A、B、E、F 根据半定量视觉分析,Bapi-AD患者的AP密度与NI-AD患者相似 (
图1C和D
和
图2C和D
) ,与匹配 APOE公司 基因型。 在3名Bapi-AD受试者中,没有肉眼可见的广泛AP切除(斑片状或其他)的组织学证据。 此外,经硫黄烷-S染色的大脑皮层孤立血管网中的CAA密度显示,所有3例患者都有中度至重度淀粉样蛋白沉积(
图1H
,
2G和2H ). 经硫黄素-S染色的病例2的整个软脑膜血管轴的全套组织学制剂显示,中度至重度存在以非常均匀的方式分布的血管淀粉样蛋白(
图1G ). 我们之前发布的关于Bapi-AD的报告 [14] 与本研究病例1相对应,显示出严重的软脑膜CAA(参见 图1D 参考 [14] ). 病例#3未评估脑膜Aβ。
与NDC病例相比,Bapi-AD病例#1和#2在额叶和颞叶皮质中显示出密集的小胶质细胞分布,CD68抗体染色; 然而,Bapi-AD和NI-AD患者的小胶质细胞密度没有差异 (
图3A、B和C
) 。HLA-DR抗体证实了这一观察结果 (
图3D、E和F
) .HLA-DR免疫反应激活的小胶质细胞在Bapi-AD病例#3的大脑皮层和白质内大量存在,并且在脑室下尤其是杏仁核内数量更多 (
图3G
) 例1和例2的额叶和颞叶皮质均无T细胞淋巴细胞浸润。 相反,Bapi-AD病例#3显示CD3免疫反应性T淋巴细胞稀疏但明确的浸润,通常位于血管周围位置,位于大脑皮层和白质内。 在浆膜下观察到较高浓度的T淋巴细胞,更显著的是在杏仁核内 (
图3H
) GFAP免疫荧光染色显示纤维状星形胶质细胞 (
图4A
) .NI-AD病例 (
图4B
) 与Bapi-AD组相比,似乎有更大体积的星形细胞细胞质和更频繁的突起 (
图4C
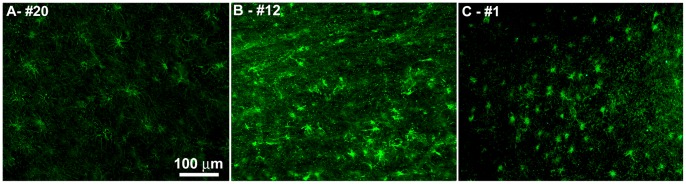
) 尽管Bapi-AD患者的颞叶中促炎细胞因子TNF-α水平高于NI-AD队列,但我们并未观察到颞叶小胶质细胞形态或密度的相应变化。
图3。 免疫组化的典型图像显示小胶质细胞和T淋巴细胞。
A) 21号NDC病例颞皮质小胶质细胞CD68染色。 B) NI-AD病例#11颞叶皮质CD68染色。 C) Bapi-AD病例#2颞叶皮质CD68染色。 D) 21号NDC病例额叶皮质小胶质细胞HLA-DR染色。 E) NI-AD病例#11颞叶皮质HLA-DR染色。 F) Bapi-AD病例#2颞叶皮质HLA-DR染色。 G) Bapi-AD病例#3颞叶皮质HLA-DR染色。 H) Bapi-AD病例#3颞叶皮质T淋巴细胞CD3染色。 放大倍数: A–F –200X和 G公司 , H(H) –100倍。
图4。 GFAP免疫荧光染色的代表性图像。
A) NDC病例#20的额叶皮层。 B) NI-AD病例#12的颞叶皮层。 C) Bapi-AD病例#1的颞皮质。 有关更多详细信息,请参阅结果部分。 放大倍数: A–C –200倍。
Aβ的量化 40 酶联免疫吸附试验显示,在额叶和颞叶 (
图5A和5B
) Bapi-AD患者的Tris-soluble分数平均比NI-AD患者分别增加了18倍和32倍,尽管只有额叶水平 (
图5A
) 达到统计显著性( 第页 <0.05). 在颞叶,可溶性Aβ 40 由于Bapi-AD值的扩散,没有统计学意义 (
图5B
) 相比之下,NI-AD组的Tris-soluble Aβ含量仅增加了2.0倍(额叶)和2.7倍(颞叶) 40 与国家数据中心队列相比 (
图5A和5B
) 对于Tris-提取的Aβ,观察到类似但不那么引人注目的情况 42 NI-AD和Bapi-AD患者额叶和颞叶的平均水平分别增加了2.3倍和1.4倍 (
图5E和5F
) .
图5。 额叶和颞叶中可溶性和不溶性Aβ的ELISA定量。
A) 额叶皮层三联可溶性Aβ 40 注意NI-AD和Bapi-AD的Aβ水平之间的显著差异。 B) 颞皮质三可溶性Aβ 40 NI-AD和Bapi-AD之间存在较大的平均差异,但由于数值的扩散,没有统计学上的显著差异。 正面( C类 )和时间( D类 )皮质GDFA/GHCl-可溶性Aβ 40 在这两种情况下,平均Aβ均显著升高 40 在Bapi AD中相对于NI-AD。正面( E类 )和时间( F类 )皮质三联可溶性Aβ 42 在两个肺叶中,由于数值的扩散,NI-AD和Bapi-AD之间没有统计差异。 正面( G公司 )和时间( H(H) )皮质GDFA/GHCl-可溶性Aβ 42 Bapi-AD aβ平均下降 42 相对于NI-AD,后者比前者更明显。 Bapi-AD列(1、2和3)中的数字对应于
表1 使用单向方差分析和Tukey多重比较测试进行统计分析(* 第页 = 0.05–0.01; ** 第页 = 0.01–0.001; *** 第页 <0.0001). 缩写:Tris,20mM Tris-HCl,5mM EDTA,pH 7.8,加蛋白酶抑制剂混合物; GDFA,玻璃蒸馏甲酸; GHCl,5 M盐酸胍,50 mM Tris-HCl,pH 8.0,加蛋白酶抑制剂鸡尾酒; NDC,非痴呆控制; NI-AD,非免疫性阿尔茨海默病; Bapi-AD、bapineuzumab免疫性阿尔茨海默病。
Bapi-AD组和NI-AD组之间的比较表明,GDFA/GHCl固溶纤维Aβ的去除具有明显的选择性 42 , (
图5G和5H
) 伴随着不溶性纤维aβ的增加 40 (
图5C和5D
) 在额叶,GDFA/GHCl可溶性Aβ的平均值 42 Bapi-AD组总蛋白水平为429 ng/mg,而NI-AD组的平均水平与NI-AD小组的549 ng/mg总蛋白相比,免疫病例减少了1.3倍 (
图5G
) 相反,GDFA/GHCl可溶性Aβ的平均值 40 与NI-AD受试者相比,Bapi-AD组的水平升高了18倍(分别为113 ng/mg和6.2 ng/mg) (
图5C
) 在颞叶,GDFA/GHCl可溶性aβ的相对损失更明显 42 很明显,从NI-AD病例的461 ng/mg降至Bapi-AD病例平均231 ng/mm,减少了2倍 (
图5H
) 平均GDFA/GHCl可溶性时间Aβ 40 NI-AD组的水平从5.0 ng/mg增加到Bapi-AD组平均43 ng/mg,增加了8.6倍 (
图5D
) .
值得注意的是,Aβ 40 Aβ 42 与NI-AD队列相比,Bapi-AD受试者中的GDFA/GHCl可溶性总Aβ水平没有显著的净变化 (
图6C和6D
) 另一方面,与NI-AD组相比,Bapi-AD组额叶皮质中Tris-soluble总Aβ水平显著升高 (
图6A
) 只有Bapi-AD病例#3的颞叶Tris-soluble总Aβ水平高于其他2名Bapi-AD受试者 (
图6B
) Aβ的这种深刻变化 40 在Bapi-AD队列中,Aβ 42 :Aβ 40 免疫治疗后,比率发生了显著变化。
图6。 总水平(Aβ 40 +Aβ 42 )额叶和颞叶的可溶性(Tris)和不溶性(GDFA/GHCl-soluble)Aβ。
A类 )额叶皮层总三可溶性Aβ。 B) 颞叶皮层总Tris-soluble Aβ。 C类 )额叶皮层总GDFA/GHCl可溶性Aβ。 D类 )颞皮质总GDFA/GHCl可溶性Aβ。 有关统计处理和缩写,请参见图例 图5 .
对于额叶和颞叶,平均Aβ 42 :Aβ 40 NI-AD组GDFA/GHCl可溶部分的比率分别为107和112。 在Bapi-AD病例中,这些比率分别显著降至5.0和5.5 (
表2
) 。类似地,如所示
表2 ,Tris可溶平均值Aβ 42 :Aβ 40 与NI-AD病例相比,免疫病例的比率显著降低(Bapi-AD=0.31 vs.NI-AD=2.51; 8.1倍)在额叶和颞叶(Bapi AD=0.35 vs.NI-AD=2.19; 6.3倍)。
表2。 Aβ比率 42 :Aβ 40 根据ELISA测定,NI-AD和Bapi-AD受试者颞叶额叶中。
正面 世俗的
NI-AD公司 三溶性Aβ比率 42 :Aβ 40 GDFA/GHCl可溶性Aβ比率 42 :Aβ 40 NI-AD公司 三溶性Aβ比率 42 :Aβ 40 GDFA/GHCl可溶性Aβ比率 42 :Aβ 40
10 2.73 131 10 3.14 185
11 1.84 66 11 1.83 87
12 2.19 93 12 1.65 101
13 3.28 137 13 2.15 75
平均值
2.51
107
平均值
2.19
112
Bapi-AD公司
三溶性Aβ比率 42 :Aβ 40
GDFA/GHCl比率- 可溶性Aβ 42 :Aβ 40
Bapi-AD公司
三溶性Aβ比率 42 :Aβ 40
GDFA/GHCl可溶性Aβ比率 42 :Aβ 40
1 0.154 2 1 0.67 5.36
2 0.163 5.11 2 0.35 6.39
三 0.608 7.93 三 0.04 4.78
平均值
0.308
5.01
平均值
0.35
5.51
* 第页 =
0.0025
0.0037
0.0073
0.0153
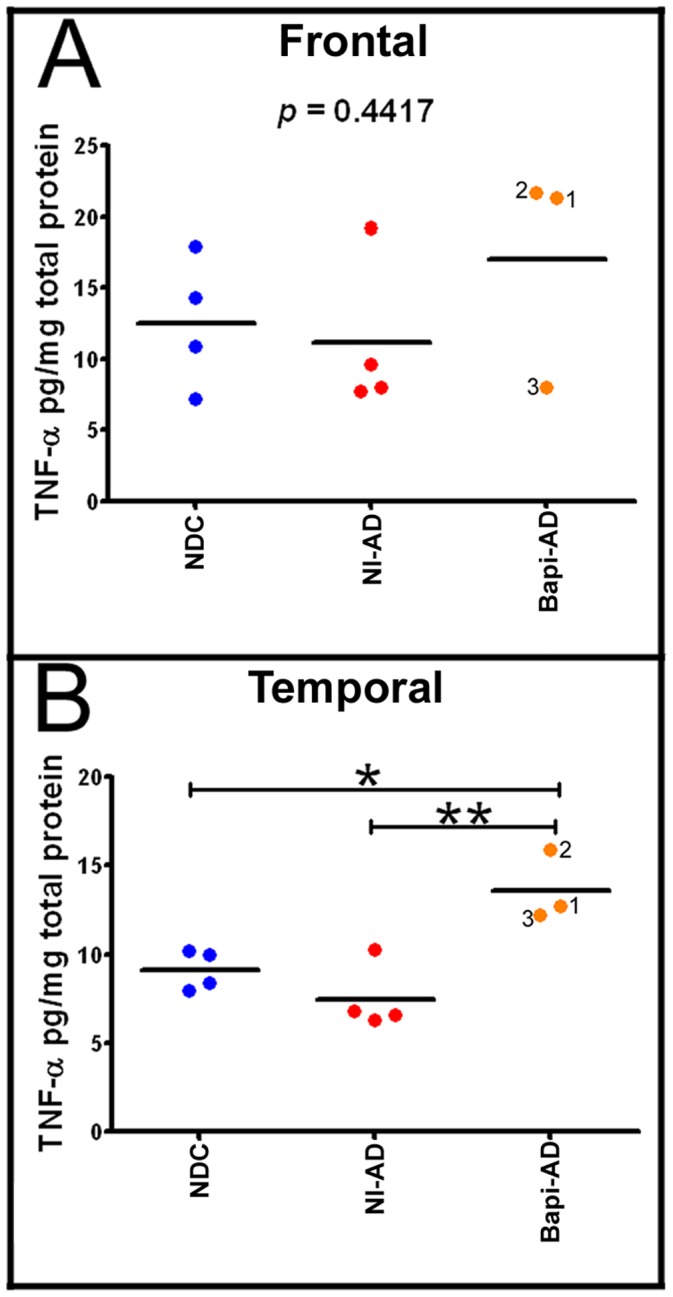
与NI-AD患者相比,Bapi-AD患者额叶和颞叶的促炎性TNF-α分子平均增加,但这种差异仅在颞叶达到统计显著性水平 (
图7A和7B
) 如上所述,与NI-AD组相比,Bapi-AD组的Aβ在颞叶的去除更广泛,这可以解释这种促炎细胞因子的相对增加。
图7。 ELISA定量肿瘤坏死因子-α(TNF-α)。
A类 )额叶皮层TNF-α水平和 B类 )颞叶皮层TNF-α水平。 注意,与NI-AD相比,Bapi AD颞叶中这种细胞因子的数量显著增加。有关统计处理和缩写,请参阅图例 图5 .
使用CT20AβPP抗体通过Western blot对AβPP含量进行评估,结果显示Bapi-AD和NI-AD患者的额叶和颞叶没有统计学上的显著差异 (
图8A和8B
) CT20AβPP抗体还检测CT99(~11 kDa)和CT83(~9 kDa (
图8A和8B
) 分别通过β-和α-分泌酶从AβPP蛋白水解中获得的肽。 与NDC相比,NI-AD额叶中的这些肽显著增加 (
图8A
) ,但与Bapi-AD组无差异。 HT7抗体对总τ的鉴定表明,亚型的预期多样性范围为~58~~40 kDa。 然而,这些亚型的总量在研究组或大脑区域之间没有显示出统计差异 (
图8C和8D
) .
图8。 AβPP及其C末端肽CT99、CT83和τ亚型的Western blot分析。
正面( A类 )和时间( B类 )CT20AβPP的皮质Western blots。 针对AβPP的最后20个氨基酸制备的CT20AβPP抗体用于检测AβPP及其CT肽。 正面( C类 )和时间( D类 )皮质τ的西方斑点。 使用针对tau分子159-163氨基酸的HT7克隆来探测tau亚型。 分子量,以kDa为单位,在每个印迹的左侧给出。 肌动蛋白再probes如下所示,作为总蛋白负荷控制。 为了进行比较,对同一凝胶中的NDC和NI-AD进行了所有蛋白质印迹分析。 免疫组和非免疫组之间没有统计学差异。 图例中描述了统计分析和缩写 图5 .
讨论 我们的调查显示: 1) 根据组织学和数量,与年龄和 APOE公司 基因型匹配的NI-AD病例; 2) 与NDC队列相比,Bapi-AD和NI-AD病例中的微胶质增生和星形胶质增生增加,而T淋巴细胞浸润仅出现在Bapi-AD#3病例中; 3) 在Bapi-AD受试者中,Aβ的总量 42 相对于Aβ减少 40 水平随之升高; 4) Aβ显著降低 42 :Aβ 40 Bapi-AD和NI-AD队列之间的比率; 和 5) 尽管Aβ发生了变化,但Bapi-AD和NI-AD之间的AβPP、CT99和CT83的相对水平没有明显差异 42 :Aβ 40 比率。 目前尚不清楚免疫治疗后Aβ肽组分的变化是否代表了对AP和CAA解聚的真正的代偿性生理反应,还是仅仅反映了在变化的环境条件下由于AβPP的连续加工而产生的新平衡。 有趣的是,与NI-AD患者中观察到的水平相比,Bapi-AD患者的Tris-soluble Aβ池明显增加。 无法离开大脑的可溶性Aβ水平升高的病理生理学后果尚不清楚。
根据每次输注的抗体量和相对于Aβ肽水平的给药剂量,对巴匹纽珠单抗的个体反应进行详细检查,并没有显示额叶和颞叶中Tris可溶性Aβ组分的明确模式。 然而,在Bapi-AD个体中,GDFA/GHCl-可溶性Aβ的水平 40 和Aβ 42 组分有聚集在一起的趋势。
Germane是我们之前在Aβ中观察到的类似变化 42 :Aβ 40 主动免疫AN-1792(含Aβ 42 肽)疫苗与6名NI-AD受试者进行比较 [5] .GDFA/GHCl提取的Aβ的ELISA也显示Aβ的平均相对损失 42 以及aβ的伴随平均增加 40 在AN-1792受试者中。 平均Aβ 42 :Aβ 40 NI-AD和AN-1792受试者的比率分别为17.8和0.59,因此显示出类似于当前bapineuzumab研究中所显示的趋势。 作为一组,AN-1792免疫受试者的GDFA/GHCl可溶性总aβ平均损失为9.3%。 此外,与NI-AD相比,AN-1792受试者的总三溶性Aβ升高了19倍 [5] .
Aβ中的表面优先耗竭 42 bapineuzumab免疫治疗可能反映了抗体对aβ特定构象域的亲和力 42 肽。 Aβ的分子模拟 42 表明1Asp的N-末端氨基和Aβ的C-末端羧基之间存在离子键 42 阿拉 [24] 二聚体Aβ表面 42 这种分子内构象可能对抗原-抗体相互作用具有较高的亲和力。 从丝状结构中去除二聚体Aβ很可能是抗体从AP中分解这些结构的方式,因为二聚体中的分子间键在热力学上比处于错误折叠状态的自由亚稳单体中的键稳定十亿倍 [25] – [27] 此外,bapineuzumab免疫治疗的效果可能有限,因为由于这些分子的部分N末端降解,在沉积的aβ肽中,其特定表位靶点部分或全部缺失 [28] , [29] 以及翻译后修饰,如天冬氨酸同体化 [28] 和焦戊酰环化 [30] ,增强聚合和二聚体对酶降解的抵抗力 [31] 相反,在AN-1792免疫个体中观察到的明显更完全分解的斑块骨架 [9] 可能是该疫苗的多克隆特性的结果,该疫苗可能有效地消除了aβ 42 以及广泛的其他物种,包括N-末端降解的aβ肽。 如果导致痴呆的一连串有害事件确实伴随淀粉样蛋白沉积,则表明aβ和 40 和Aβ 42 必须先发制人和彻底。
重要的是要记住,尽管bapineuzumab免疫治疗影响了Aβ 42 水平,它并没有明显抑制淀粉样蛋白AβPP加工或沉积的主要动力。 Aβ的损失 42 Aβ显著升高 40 提出一种可能的代偿机制,其中aβ的一种形式被另一种形式取代。 例如,Aβ的代偿沉积 40 可能解释了Bapi-AD和NI-AD患者尽管存在Aβ 42 消耗。 在Aβ免疫个体中,明显的代偿性生成和观察到的高水平血管淀粉样蛋白支持了这样一种假设,即Aβ肽的功能之一是生成一个保护性止血样斑块/痂,以封闭大脑微血管开窍侧的泄漏血管(见 [32] – [35] ). 巴匹纽单抗免疫疗法诱导AD患者脑渗出性血管源性水肿,定义为淀粉样蛋白相关成像异常(ARIA-E)和微出血(ARIA-H) [36] Flair-MRI扫描显示,210名接受巴匹诺单抗治疗的AD患者中有36名(17%)出现血管源性水肿,其中17名(47%)出现微出血,而其余177名无ARIA-E的患者中只有7名(4%)出现微血 [37] 在Bapi-AD受试者的其他研究中观察到短暂的血管源性水肿 [12] , [38] , [39] 一项对2762名AD患者的免疫治疗临床试验基线研究发现,AD相关血管源性水肿罕见,仅发生在2例患者中 [40] 免疫AβPP Tg小鼠产生微出血是一个有充分记录的观察结果 [41] – [46] 血脑屏障(BBB)破裂可能是由bapineuzumab或高水平Aβ产生的自身抗体以及血管炎症反应引起的。 综上所述,这些观察结果表明Aβ的部分去除 42 bapineuzumab免疫治疗导致aβ生成增加的循环 40 试图在免疫治疗引起的改变面前保持BBB的完整性。
有趣的是,在显性遗传阿尔茨海默病网络(DIAN)研究中,6%的年轻无症状FAD突变携带者出现脑微出血,25%的轻度症状携带者也有这些病变,其中16%有1-4个,9%有>5个微出血 [47] 此外,纯PS Tg小鼠没有AβPP突变,因此没有AP,表现出广泛的超微结构微血管病理学 [48] 年龄较大的AβPP/PS1 Tg小鼠(19-23个月龄)在没有免疫治疗的情况下,在血管淀粉样蛋白沉积附近的区域也出现了自发的ARIA-E和ARIA-H [49] , [50] .
随着大脑老化,不可避免的微血管减少和累积损伤会降低血脑屏障的完整性 [51] .高血压、低血压、糖尿病、动脉粥样硬化、动脉硬化等慢性疾病 [52] , [53] 以及脑外伤 [54] , [55] 对脑微循环有有害影响 [56] – [59] 猪部分结扎胸主动脉会导致高血压和aβ大量增加 40, Aβ 42 和p 412 -这些动物大脑中的tau [60] ,表明全身灌注衰竭与AD相关的关键病理级联协同作用。淀粉样纤维的物理属性,如化学稳定性、不溶性、两亲性结构、水泥样性质(综述于 [32] )、金属和血红素结合能力 [24] , [61] – [64] 淀粉样蛋白能够与细胞外基质相互作用,生成弹性网状物,类似于凝血级联反应,使淀粉样蛋白成为理想的脑微血管修复/保护物质 [32] , [33] , [65] 此外,Aβ结合并隔离正常情况下被排除在大脑之外的血浆分子 [66] – [71] Aβ肽还与凝血酶、纤维蛋白原和纤溶酶原等关键凝血分子相互作用 [72] – [78] 在这种情况下,AβPP 770 和AβPP 751 含有Kunitz蛋白酶抑制剂结构域的亚型在凝血级联中起着重要的调节作用 [79] , [80] 此外,在人类中,复杂的淀粉样沉积物似乎受到α1-抗糜蛋白酶的蛋白水解降解的保护 [81] , [82] 脑微血管周围淀粉样蛋白的持续过度增生可能会通过收缩管腔、造成毛细血管残端和灌注受损而终止 [32] 可以假设轻度和中度AD患者的大脑已经达到或正在接近适应性平衡,其中淀粉样蛋白的生成几乎达到了一个平台 [83] 在这些情况下,Aβ的去除 42 可能会触发aβ的补偿性过度生产 40 .
尽管大脑某些区域的AP降低,但抗淀粉样蛋白免疫治疗 [5] – [9] , [84] 和/或淀粉样蛋白组成的显著改变,一直未能在痴呆的临床过程中产生相应的变化 [7] , [10] , [11] 虽然波尼祖单抗、巴匹诺珠单抗和索拉奈珠单抗治疗轻度和中度AD的临床试验已经停止或没有达到其主要终点 [11] , [85] , [86] ,其他使用各种免疫方法的临床试验仍在评估中。 抗Aβ免疫疗法可显著增加可溶性Aβ肽,这一观察结果可能解释了这些干预措施对痴呆症的微弱影响。 从AD脑中分离出可溶性/低聚物Aβ分子 [87] 这些肽显然具有神经毒性 [88] , [89] 此外,这些物种在很大程度上被证明在AD的发病机制和病理生理演变中发挥着核心作用,并在一定程度上取代了不溶性斑块和血管aβ作为治疗干预的主要靶点。 如果这些原则是正确的,并且考虑到目前的证据,预计在免疫接种的AD患者中,可溶性低聚物Aβ的增加会加重疾病的自然进程。 有待进行更大的研究,以更准确地评估可溶性/低聚物Aβ在免疫治疗受者中所起的作用。
阿尔茨海默病预防(临床前)倡议(API)和DIAN研究将涉及在认知功能障碍出现的估计时间前几年,对患有PS突变的受试者主动给予抗Aβ免疫治疗。 临床试验证实,抗淀粉样蛋白免疫治疗对散发性AD的关键生物标志物产生了一系列显著的积极和消极影响 [5] – [9] , [12] , [14] , [39] , [84] , [90] – [95] 然而,在PS突变携带者中,策略性定时的症状前治疗是否会产生类似的反应和/或保持认知功能尚不确定。 PS突变的生物化学研究导致了广泛持有的假说,即Aβ的生成丰富 42 痴呆症的基础 [96] , [97] ,尽管对FAD患者的Aβ肽进行严格定量研究表明,这一规律显然并不普遍 [98] , [99] 无论是crenezumab、solanezumab还是gangenerumab治疗都有利于Aβ 40 FAD-PS突变携带者、API和DIAN的产生和积累将提供一个理想的机会来直接测试Aβ过量的假设 42 仅生产就特别引发痴呆症的出现。 然而,一些证据,包括免疫治疗试验的临床结果令人失望,但信息量很大,表明痴呆进展具有未经认可的生化复杂性。 我们对免疫治疗的分子后果的研究和以前的研究 [5] , [9] , [14] 表明Aβ 40 可能不是完全良性的,因为该物种数量的增加与血管淀粉样变的增加和痴呆症的持续下降密切相关。 此外,API和DIAN将提供一个理想的前景,以明确检查这些特定生物标记物与痴呆发病和进展的潜在关联,并测试淀粉样级联假说的有效性。
总之,我们的研究和先前的研究证实,Aβ免疫疗法对主要AP和Aβ42靶点有深刻影响。 在某些情况下,免疫治疗导致AP的物理破坏。 在我们的研究中,尽管AP在生理上没有改变,但受试者的淀粉样蛋白谱受到Aβ水平的根本干扰 42 显著降低和Aβ 40 水平增加。 尽管有这些反应,免疫治疗并没有对痴呆症的发展产生任何实质性影响。 这种平衡转移的确切机制尚不清楚,但免疫治疗的净效应可能是较短且更可溶性的aβ的显著优先积累 40 物种。 再加上公认的AD生物化学和神经病理学的复杂性,这些观察结果表明,淀粉样斑块沉积物要么不是认知障碍的唯一诱因,要么其精确的分子结构对痴呆症的出现几乎没有总体影响。 如果是这样的话,它表明尽管对AD生物标记物有显著影响,但痴呆免疫治疗持续缺乏成功反映出普遍未能全面了解淀粉样蛋白沉积的功能及其动力学。
致谢 我们对道格拉斯·沃克博士的表演表示感谢 APOE公司 基因分型,并向Walter M.Kalback博士和Dean C.Luehrs博士提交对手稿的批判性审查。
资金筹措表 本研究得到了国家老龄研究所(NIA)拨款R01 AG-19795、NIA亚利桑那州老年痴呆症核心中心P30 AG-19610、亚利桑那州老年痴呆研究联合会以及加利福尼亚大学(欧文)ADRC NIH/NIA拨款P50 AG-16573的支持。 Banner Sun Health Research Institute的脑部捐赠计划得到了国家神经疾病和中风研究所(U24 NS072026)、亚利桑那州卫生服务部(合同211002,亚利桑那州阿尔茨海默病研究中心)、, 亚利桑那州生物医学研究委员会(与亚利桑那州帕金森病联盟签订4001001005-901和1001号合同)和迈克尔·福克斯帕金森病研究基金会。 资助者在研究设计、数据收集和分析、决定出版或准备手稿方面没有任何作用。
工具书类
1 Ferri CP、Prince M、Brayne C、Brodaty H、Fratiglioni L等(2005)《全球痴呆流行率:德尔菲共识研究》。 柳叶刀 366: 2112–2117. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
2 Pul R、Dodel R、Stangel M(2011),阿尔茨海默病的抗体治疗。 生物治疗专家 11: 343–357. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
三。 Morgan D(2011)阿尔茨海默病免疫治疗。 内科学杂志 269: 54–63. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
4 Kerchner GA,Boxer AL(2010)Bapineuzumab。 生物治疗专家 10: 1121–1130. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
5 Maarouf CL、Daugs ID、Kokjohn TA、Kalback WM、Patton RL等(2010)抗淀粉样蛋白免疫治疗的生化后果。 摩尔神经变性剂 5: 39. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
6 Boche D、Zotova E、Weller RO、Love S、Neal JW等(2008)阿贝塔免疫对人类阿尔茨海默病大脑血管系统的影响。 大脑 131: 3299–3310. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
7 Holmes C、Boche D、Wilkinson D、Yadegarfar G、Hopkins V等(2008)阿贝塔42免疫对阿尔茨海默病的长期影响:随机安慰剂对照一期试验的随访。 柳叶刀 372: 216–223. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
8 Serrano-Pozo A、William CM、Ferrer I、Uro-Coste E、Delisle MB等(2010年),人类抗淀粉样β主动免疫对神经突起形态和tau病理学的有益影响。 大脑 133: 1312–1327. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
9 Patton RL、Kalback WM、Esh CL、Kokjohn TA、Van Vickle GD等(2006),AN-1792免疫性阿尔茨海默病患者的淀粉样β肽残留:一项生化分析。 美国病理学杂志 169: 1048–1063. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
10 Gilman S、Koller M、Black RS、Jenkins L、Griffith SG等(2005年)阿贝塔免疫(AN1792)对AD患者的临床影响。 神经病学 64: 1553–1562. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
11 Fagan T(2012)静脉注射Bapineuzumab的临床试验。 可用: http://www.alzforum.org/new/detail.asp?id=3234 。访问日期:2012年10月15日。
12 Salloway S、Sperling R、Gilman S、Fox NC、Blennow K等(2009年)巴匹诺单抗治疗轻中度阿尔茨海默病的2期多剂量递增试验。 神经病学 73: 2061–2070. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
13 Beach TG、Sue LI、Walker DG、Roher AE、Lue L等(2008)《太阳健康研究所脑捐赠计划:描述和经验》,1987年至2007年。 细胞组织库 9: 229–245. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
14 Roher AE、Maarouf CL、Daugs ID、Kokjohn TA、Hunter JM等(2011年),Bapineuzumab免疫治疗受者的神经病理学和Aβ谱。 阿尔茨海默病杂志 24: 315–325. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
15 Mirra SS(1997)CERAD神经病理学方案和阿尔茨海默病尸检诊断的一致建议:评论。 神经生物老化 18:S91–S94。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
16 Braak H,Braak E(1991)阿尔茨海默病相关变化的神经病理分期。 神经病理学学报 82: 239–259. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
17 Sabbagh MN、Cooper K、DeLange J、Stoehr JD、Thind K等(2010)功能性、整体性和认知性衰退与MCI和AD中阿尔茨海默病病理学的积累相关 7: 280–286. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
18 Roher AE、Esh C、Kokjohn TA、Kalback W、Luehrs DC等(2003)Willis动脉粥样硬化圈是散发性阿尔茨海默病的危险因素。 动脉硬化血栓血管生物 23: 2055–2062. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
19 美国国家老龄研究所和里根研究所阿尔茨海默病神经病理学评估诊断标准工作组(1997年)对阿尔茨海默氏病尸检诊断的一致建议。 神经生物老化 18:S1–S2。 [ 公共医学 ] [ 谷歌学者 ]
20 Head E,Azizeh BY,Lott IT,Tenner AJ,Cotman CW,et al.(2001)唐氏综合征老年人大脑中神经元和β-淀粉样蛋白沉积的补体相关性。 神经生物学疾病 8: 252–265. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
21 Saing T、Dick M、Nelson PT、Kim RC、Cribbs DH等(2012),拳击性痴呆患者的额叶皮层神经病理学。 神经创伤杂志 29: 1054–1070. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
22 Head E、Starr A、Kim RC、Parachikova A、Lopez GE等人(2006)以路易体痴呆为特征的复发性多软骨炎。 神经病理学学报 112: 217–225. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
23 Maarouf CL、Daugs ID、Kokjohn TA、Walker DG、Hunter JM等(2011)阿尔茨海默病和非痴呆高病理控制非老年人:比较和对比认知成功衰老的生物化学。 公共科学图书馆一号 6:e27291。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
24 Chaney MO、Webster SD、Kuo YM、Roher AE(1998)《阿尔茨海默病Abeta1-42肽的分子模型》。 蛋白质工程 11: 761–767. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
25 Zhang Y,Kim B-H,Yu J,Lovas S,Lyubchenko Y(2012)β-淀粉样蛋白错误折叠和聚集的分子机制:实验和理论的见解。 老年痴呆症 第8页:第659页。 [ 谷歌学者 ]
26 Kim BH、Palermo NY、Lovas S、Zaikova T、Keana JF等(2011)Abeta-40相互作用的单分子原子力显微镜力谱研究。 生物化学 50: 5154–5162. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
27 Lyubchenko YL、Kim BH、Krasnoslobodtsev AV、Yu J(2010)《蛋白质错误折叠疾病的纳米成像》。 Wiley Interdiscip Rev纳米生物技术 2: 526–543. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
28 Roher AE、Lowenson JD、Clarke S、Wolkow C、Wang R等(1993)β-淀粉样蛋白核心蛋白肽链的结构改变可能是其在阿尔茨海默病中沉积和稳定的原因。 生物化学杂志 268: 3072–3083. [ 公共医学 ] [ 谷歌学者 ]
29 Gowing E、Roher AE、Woods AS、Cotter RJ、Chaney M等(1994)Aβ17-42肽的化学表征,Aβ17~42肽是阿尔茨海默病弥漫性淀粉样沉积的一种成分。 生物化学杂志 269: 10987–10990. [ 公共医学 ] [ 谷歌学者 ]
30 Kuo YM、Emmerling MR、Woods AS、Cotter RJ、Roher AE(1997年),从神经炎斑块和血管淀粉样沉积中分离、化学表征和定量β3-焦氨酰肽。 生物化学-生物物理研究委员会 237: 188–191. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
31 Kuo YM,Webster S,Emmerling MR,De LN,Roher AE(1998)不可逆二聚化/四聚化和翻译后修饰抑制阿尔茨海默病Aβ肽的蛋白水解降解。 Biochim生物物理学报 1406: 291–298. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
32 Kokjohn TA、Maarouf CL、Roher AE(2012)阿尔茨海默病淀粉样变是修复机制误入歧途的结果吗? 老年痴呆症 8: 574–583. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
33 Atwood CS,Bowen RL,Smith MA,Perry G(2003)脑血管对维持血管完整性和血液供应的密封剂、抗凝血剂和重塑分子的需求。 大脑研究大脑研究进展 43: 164–178. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
34 Cullen KM,Kocsi Z,Stone J(2006),老龄人脑的微血管病理学:老年斑块是微出血部位的证据。 神经生物老化 27: 1786–1796. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
35 Cullen KM,Kocsi Z,Stone J(2005),毛细血管周围富含血液的沉积物:衰老人类大脑皮层微出血的证据。 大脑血流代谢杂志 25: 1656–1667. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
36 Sperling RA、Jack CR Jr、Black SE、Frosch MP、Greenberg SM等(2011),淀粉样蛋白修饰治疗试验中淀粉样蛋白相关成像异常:阿尔茨海默病协会研究圆桌工作组的建议。 老年痴呆症 7: 367–385. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
37 Sperling R、Salloway S、Brooks DJ、Tampieri D、Barakos J等(2012年),使用bapineuzumab治疗阿尔茨海默病患者的淀粉样蛋白相关成像异常:回顾性分析。 柳叶刀神经病学 11: 241–249. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
38 Black RS、Sperling RA、Safirstein B、Motter RN、Pallay A等(2010年),对阿尔茨海默病患者进行的bapineuzumab单剂量递增研究。 阿尔茨海默病相关疾病 24: 198–203. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
39 Rinne JO、Brooks DJ、Rossor MN、Fox NC、Bullock R等人(2010)11C PiB PET评估巴匹纽单抗治疗的阿尔茨海默病患者纤维淀粉样蛋白β负荷的变化:一项2期、双盲、安慰剂对照、递增剂量的研究。 柳叶刀神经病学 9: 363–372. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
40 Carlson C、Estergard W、Oh J、Suhy J、Jack CR Jr等(2011年),赛马司他和索拉奈珠单抗3期临床试验前阿尔茨海默病研究队列中无症状血管源性水肿的患病率。 老年痴呆症 7: 396–401. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
41 Pfeifer M、Boncristiano S、Bondolfi L、Stalder A、Deller T等(2002),被动抗阿贝塔免疫治疗后的脑出血。 科学类 298: 1379. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
42 Wilcock DM、Jantzen PT、Li Q、Morgan D、Gordon MN(2007)淀粉样β疫苗接种,但非硝基非甾体抗炎药治疗,会增加血管淀粉样蛋白和微出血,同时两者都会减少实质淀粉样蛋白。 神经科学 144: 950–960. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
43 Wilcock DM、Rojiani A、Rosenthal A、Subbarao S、Freeman MJ等(2004年)。尽管血管淀粉样蛋白和微出血增加,但老龄APP-转基因小鼠对阿贝塔的被动免疫治疗可逆转认知缺陷并消耗实质淀粉样蛋白沉积。 神经炎症杂志 1: 24. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
44 Racke MM,Boone LI,Hepburn DL,Parsadainian M,Bryan MT,et al.(2005)免疫治疗对淀粉样前体蛋白转基因小鼠脑淀粉样血管病相关微出血的加剧依赖于对淀粉样β沉积形式的抗体识别。 神经科学 25: 629–636. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
45 Kumar-Singh S、Pirici D、McGowan E、Serneels S、Ceuterick C等(2005),老年痴呆症Tg2576和PSAPP小鼠模型中的致密核斑块集中在血管壁上。 美国病理学杂志 167: 527–543. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
46 Luo F、Rustay NR、Seifert T、Roesner B、Hradil V等(2010),淀粉样前体蛋白转基因小鼠被动免疫治疗期间脑微出血的磁共振成像检测和时间进程。 药理学实验与治疗杂志 335: 580–588. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
47 Strobel G(2012)扩展网络,DIAN开始显示纵向数据。 可用: http://www.alzforum.org/new/detail.asp?id=3290 。2012年10月15日查阅。
48 Gama Sosa MA、Gasperi RD、Rocher AB、Wang AC、Janssen WG等(2010)表达早老素1相关家族性阿尔茨海默病突变的转基因小鼠的年龄相关血管病理学。 美国病理学杂志 176: 353–368. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
49. Goodman J,Freeman G,Angus W,Brown T,Cheng-te Chou P,et al.(2012)老年APP+早老蛋白1小鼠微出血和渗出/水肿类型的自发性淀粉样蛋白相关成像异常。 老年痴呆症 8:第11页至第12页。 [ 谷歌学者 ]
50 Goodman J、Freeman G、Angus W、Brown T、Cheng te Chou P等人(2012)老年APP+PS1小鼠的自发性ARIA-E和ARIA-H。 老年痴呆症 第8页:第153页。 [ 谷歌学者 ]
51 Zlokovic BV(2011)阿尔茨海默病和其他疾病中神经变性的神经血管途径。 Nat Rev神经科学 12: 723–738. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
52 Kalaria RN(2010)《大脑退化的血管基础:痴呆症的摇晃控制和风险因素》。 螺母版本 68补遗2S74–S87。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
53 Craft S(2009)《代谢紊乱在阿尔茨海默病和血管性痴呆中的作用:两条道路交汇》。 神经系统科学 66: 300–305. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
54 Nag S,Kapadia A,Stewart DJ(2011)综述:急性脑损伤血脑屏障破坏的分子发病机制。 神经病理学应用神经生理学 37: 3–23. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
55 Shlosberg D、Benifla M、Kaufer D、Friedman A(2010),血脑屏障破坏是创伤性脑损伤的治疗靶点。 Nat Rev Neurol公司 6: 393–403. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
56 Kalaria RN、Akinyemi R、Ihara M(2012)血管病理学是否会导致阿尔茨海默病改变? 神经科学杂志 1322: 141–147. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
57 Hachinski V、Kalaria RN、Deramecourt V(2012),痴呆患者脑血管病变的分期和自然病史。 神经病学 79: 107. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
58 de la Torre JC(2012)《脑血流动力学和血管风险因素:为阿尔茨海默病奠定基础》。 阿尔茨海默病杂志。 [ 内政部 ] [ 公共医学 ]
59 Dickstein DL、Walsh J、Brautigam H、Stockton SD Jr、Gandy S等(2010),血管危险因素和血管功能障碍在阿尔茨海默病中的作用。 西奈山医学院 77: 82–102. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
60 Kuo YM,Shih YH,Lee CW,Jiang MJ,Lin PY(2012)高血压会增加猪的tau过度磷酸化和β-淀粉样蛋白生成。 老年痴呆症 第8页:第651页。 [ 谷歌学者 ]
61 Tougu V,Tiiman A,Palumaa P(2011)锌(II)和铜(II)离子与阿尔茨海默氏淀粉样β肽的相互作用。 金属离子结合,对纤维化的贡献和毒性。 金属组学 3: 250–261. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
62 Kenche VB,Barnham KJ(2011)《阿尔茨海默病与金属:治疗机会》。 杂志 163: 211–219. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
63 Chuang JY,Lee CW,Shih YH,Yang T,Yu L,et al.(2012)淀粉样β与血红蛋白之间的相互作用:阿尔茨海默病淀粉样斑块形成的意义。 公共科学图书馆一号 7:e33120。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
64 Roberts BR,Ryan TM,Bush AI,Masters CL,Duce JA(2012)金属生物学和淀粉样蛋白β肽在阿尔茨海默病中的作用。 神经化学杂志 120补充1149–166。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
65 Roher AE、Lowenson JD、Clarke S、Woods AS、Cotter RJ等(1993)β-淀粉样蛋白-(1-42)是脑血管淀粉样蛋白沉积的主要成分:对阿尔茨海默病病理学的影响。 美国国家科学院程序 90: 10836–10840. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
66 Bohrmann B、Tjernberg L、Kuner P、Poli S、Levet-Trafit B等(1999)控制淀粉样β肽聚合的内源性蛋白质。 中枢神经系统和外周组织中β-淀粉样蛋白形成的可能影响。 生物化学杂志 274: 15990–15995. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
67 Biere AL、Ostaszewski B、Stimson ER、Hyman BT、Maggio JE等人(1996)淀粉样β肽通过人血浆中的脂蛋白和白蛋白转运。 生物化学杂志 271: 32916–32922. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
68 Kuo YM、Kokjohn TA、Kalback W、Luehrs D、Galasko DR等(2000)淀粉样β肽与血浆蛋白和红细胞相互作用:对其在血浆中定量的影响。 生物化学-生物物理研究委员会 268: 750–756. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
69 Milojevic J,Melacini G(2011)人类血清白蛋白-阿尔茨海默氏阿贝塔肽相互作用的计量学和亲和力。 生物物理学J 100: 183–192. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
70 Xi G,Reiser G,Keep RF(2003)凝血酶和凝血酶受体在缺血性、出血性和创伤性脑损伤中的作用:有害还是保护? 神经化学杂志 84: 3–9. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
71 Matsuoka H,Hamada R(2002)凝血酶在与脑出血相关的中枢神经系统损伤中的作用:药物干预的机会? 中枢神经系统药物 16: 509–516. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
72 Grammas P,Ottman T,Reimann-Philipp U,Larabee J,Weigel PH(2004)损伤的脑内皮细胞释放神经毒性凝血酶。阿尔茨海默病杂志 6: 275–281. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
73 Yin X,Wright J,Wall T,Grammas P(2010)脑内皮细胞在阿尔茨海默病中合成神经毒性凝血酶。 美国病理学杂志 176: 1600–1606. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
74 Gingrich MB、Traynelis SF(2000)丝氨酸蛋白酶和脑损伤——有联系吗? 神经科学趋势 23: 399–407. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
75 Ahn HJ、Zamolodchikov D、Cortes-Canteli M、Norris EH、Glickman JF等(2010)阿尔茨海默病肽β-淀粉样蛋白与纤维蛋白原相互作用并诱导其寡聚。 美国国家科学院程序 107: 21812–21817. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
76 Melchor JP,Pawlak R,Chen Z,Strickland S(2003)组织型纤溶酶原激活物(tPA)和tPA阻滞剂在阿尔茨海默病发病机制和治疗中的可能作用。 摩尔神经科学杂志 20: 287–289. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
77 Melchor JP,Pawlak R,Strickland S(2003)组织纤溶酶原激活物-纤溶酶原蛋白水解级联加速淀粉样β(Abeta)降解并抑制Abeta诱导的神经变性。 神经科学 23: 8867–8871. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
78 Cortes-Canteli M、Paul J、Norris EH、Bronstein R、Ahn HJ等(2010)纤维蛋白原和β-淀粉样蛋白相关性改变血栓形成和纤溶:阿尔茨海默病的一个可能促发因素。 神经元 66: 695–709. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
79 Van Nostrand WE、Schmaier AH、Farrow JS、Cunningham DD(1991)血小板蛋白酶连接蛋白-2/淀粉样β蛋白前体。 可能的病理和生理功能。 Ann N Y科学院 640: 140–144. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
80 Van Nostrand WE,Schmaier AH,Wagner SL(1992)蛋白酶连接蛋白-2/淀粉样β蛋白前体作为脑抗凝剂的潜在作用。 Ann N Y科学院 674: 243–252. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
81 Abraham CR,Potter H(1989)蛋白酶抑制剂α1-抗糜蛋白酶是正常衰老和阿尔茨海默病患者大脑淀粉样物质沉积的组成部分。 医学年鉴 21: 77–81. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
82 Abraham CR、McGraw WT、Slot F、Yamin R(2000)α1-抗糜蛋白酶在体内外抑制Aβ降解。 Ann N Y科学院 920: 245–248. [ 公共医学 ] [ 谷歌学者 ]
83 Jack CR Jr、Wister HJ、Vemuri P、Weigand SD、Senjem ML等(2010年)脑β-淀粉样蛋白测定和磁共振成像萎缩都预测从轻度认知障碍到阿尔茨海默病的时间进展。 大脑 133: 3336–3348. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
84 Boche D,Denham N,Holmes C,Nicoll JA(2010),主动Abeta42免疫治疗后的神经病理学:阿尔茨海默病发病机制的影响。 神经病理学学报 120: 369–384. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
85. Fagan T(2011)辉瑞公司停止Aß抗体的开发。 可用: http://www.alzforum.org/new/detail.asp?id=2950 。2012年10月15日查阅。
86 Zakaib GD(2012)第3阶段Solanezumab试验“失败”——有一线希望吗? 可用: http://www.alzforum.org/new/detail.asp?id=3254 。2012年10月15日查阅。
87. Kuo YM、Emmerling MR、Vigo-Pelfrey C、Kasunic TC、Kirkpatrick JB等(1996),正常和阿尔茨海默病大脑中的水溶性阿贝塔(N-40,N-42)低聚物。 生物化学杂志 271: 4077–4081. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
88 Roher AE,Chaney MO,Kuo YM,Webster SD,Stine WB,et al.(1996)阿贝塔-(1–42)二聚体的形态和毒性,来源于阿尔茨海默病的神经炎和血管淀粉样沉积。 生物化学杂志 271: 20631–20635. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
89. Giulian D、Haverkamp LJ、Yu JH、Karshin W、Tom D等(1996)阿尔茨海默病斑块β-淀粉样蛋白的特定结构域引发人类小胶质细胞的神经元死亡。 神经科学 16: 6021–6037. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
90 Nicoll JA、Wilkinson D、Holmes C、Steart P、Markham H等(2003年),淀粉样β肽免疫后人类阿尔茨海默病的神经病理学:病例报告。 自然·医学 9: 448–452. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
91 Orgogozo JM、Gilman S、Dartigues JF、Laurent B、Puel M等人(2003)Abeta42免疫后AD患者亚群的亚急性脑膜脑炎。 神经病学 61: 46–54. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
92 Ferrer I、Boada RM、Sanchez Guerra ML、Rey MJ、Costa-Jussa F(2004)阿尔茨海默病淀粉样β免疫接种后脑炎的神经病理学和发病机制。 大脑病理学 14: 11–20. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
93 Masliah E、Hansen L、Adame A、Crews L、Bard F等(2005)阿贝塔疫苗在阿尔茨海默病无脑炎的情况下对斑块病理学的影响。 神经病学 64: 129–131. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
94 Kokjohn TA,Roher AE(2009),一项中断试验中AD患者的抗体反应、淀粉样β肽残留和AN-1792免疫的临床效果。 中枢神经系统神经紊乱药物靶点 8: 88–97. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
95 Blennow K,Zetterberg H,Rinne JO,Salloway S,Wei J,et al.(2012)使用Bapineuzumab进行免疫治疗对轻度至中度阿尔茨海默病患者脑脊液生物标记物水平的影响。 神经系统科学 69: 1002–1010. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
96 Lleo A、Berezovska O、Growdon JH、Hyman BT(2004)与PS-1突变相关的阿尔茨海默病的临床、病理和生化谱。 美国老年精神病学杂志 12: 146–156. [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
97 Wolfe MS(2007)当损失即获得时:早老蛋白蛋白水解功能降低导致Abeta42/Abeta40增加。 早老蛋白突变在阿尔茨海默病中的作用。 EMBO代表 8: 136–140. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
98 Maarouf CL、Daugs ID、Spina S、Vidal R、Kokjohn TA等(2008)与早老蛋白突变相关的痴呆患者的组织病理学和分子异质性。 摩尔神经变性剂 3: 20. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
99 Van Vickle GD、Esh CL、Kokjohn TA、Patton RL、Kalback WM等人(2008)Presenilin-1 280Glu–>Ala突变改变C端APP处理,产生更长的伊丽莎白肽:对阿尔茨海默病的影响。 分子医学 14: 184–194. [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]