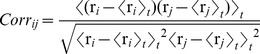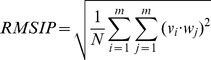摘要
PHD手指是能够解码翻译后修饰或未修饰组蛋白H3尾部的表观遗传读取器的最大家族之一。由于其直接参与人类病理学,它们越来越被视为潜在的治疗靶点。已经确定了几个PHD/组蛋白肽结构,但关于其动力学的信息相对较少。旨在描述表观遗传学读者识别组蛋白肽的动态和能量决定因素的研究将从计算研究中受益匪浅。在此,我们重点研究了PHD指亚类的动态和能量特征,该亚类专门识别K4(H3K4me0)位置未修饰的组蛋白H3肽。作为一个案例研究,我们重点研究了自身免疫调节蛋白(AIRE-PHD1)与H3K4me0复合物的第一个PHD指。自由AIRE-PHD1协方差矩阵的PCA分析强调了“扑动”运动的存在,该运动在与H3K4me0结合时被阻断在开放构象中。此外,通过分子力学/Poisson-Boltzmann表面积(MM/PBSA)方法获得的结合自由能计算结果与实验定性一致,并允许分离与AIRE-PHD1/H3K4me0配合物的天然突变体和丙氨酸突变体相关的能量项。MM/PBSA计算也已应用于识别H3K4me0的其他PHD手指的能量分析。在这种情况下,我们观察到计算的和实验的结合自由能之间有很好的相关性。总体计算表明,PHD手指识别H3K4me0依赖于静电和极性溶剂化能量项的补偿,并通过非极性相互作用稳定。
介绍
组蛋白翻译后修饰(PTM)是基因转录和DNA损伤修复等过程的重要调控平台[1]越来越多的证据表明,由于调节组蛋白PTM的修饰、安装、移除和/或解释的因素发生故障而导致的组蛋白PTMs的失调,积极地促进了人类疾病的发生和发展[2]组蛋白PTM的生物学后果通常由进化上保守的“读取器/效应器”模块介导,例如Tudor-、Bromo-和Chromo-domains,它们以修饰和上下文特异的方式与表观遗传标记结合,从而促进染色质变化或蛋白质募集[3]–[5].
在识别组蛋白H3修饰状态的专业“读取器”模块列表中,最近添加的一个模块是植物同源结构域(PHD)指。PHD手指为锌2+由50-80个氨基酸组成的结合域,形成一个两品牌的反平行β片,然后是一个α螺旋。该结构域存在于约150种人类蛋白质中,其中许多作为核小体相互作用决定因子,在组蛋白识别和表观遗传机制中起着基础作用[6]–[8]PHD模块的生理相关性通过以下基因中针对PHD手指的突变而得到强调:ING、ATRX、RAG2、和AIRE公司与发育疾病、神经和免疫疾病以及癌症有关[9]最近的结构和功能研究表明,PHD指家族可根据其对翻译后组蛋白修饰的特异性分为几个亚家族,包括组蛋白赖氨酸的甲基化状态,例如组蛋白H3赖氨酸4(H3K4me0与H3K4me2/3)、H3赖氨酸9(H3K9me3)或H3赖胺酸36(H3K 36),以及在较小程度上H3精氨酸2(H3R2me0)的甲基化状态与H3R2me2)和赖氨酸K14(H3K14)的乙酰化状态[7],[8],[10]结构表征最好的亚家族包括能够通过π-阳离子相互作用通过保守的芳族侧链配位H3K4me3/me2的PHD模块,如BPTF[11]和ING PHD手指[12],[13]BHC80的PHD模块是一个独特的子系列[14],AIRE的第一个PHD手指(AIRE-PHD1)[15],[16]CHD4的第一个和第二个PHD手指[17],装饰件24[18]和BRPF2的第一个PHD指(BRPF2-PHD1)[19],识别组蛋白H3尾部含有未修饰的赖氨酸4(H3K4me0)。在这种情况下,我们和其他人[20],[21]表明自身免疫调节蛋白AIRE(AIRE-PHD1)的第一个PHD指识别H3K4me0,从而促进其靶基因的表达。AIRE是一种主要在髓质胸腺上皮细胞(mTEC)中表达的转录激活物,它控制组织特异性抗原的表达,从而扩大诱导免疫耐受的抗原库,从而阻止自身免疫[22]最近的研究表明,AIRE通过其PHD手指模块与低甲基化H3结合,对于AIRE介导的基因表达调控和中心耐受诱导是必要的[23]重要的是AIRE公司基因[24]引起自身免疫性多内分泌病-念珠菌-表皮营养不良(APECED)[25],[26]与对BPTF和ING亚家族的观察结果不同,AIRE-PHD1不存在通过π-阳离子相互作用来协调H3K4me3的三甲基或二甲基铵离子的典型保守芳香侧链。在AIRE-PHD1中,甲基化赖氨酸结合芳香笼的关键元素被带负电荷的残基取代,这些残基可以与未甲基化的H3K4me0发生有利的相互作用,为通过芳香笼识别H3提供了一种替代方法[15],[16],[20],[21]与其他具有甲基化或非甲基化组蛋白尾部的PHD手指的观察结果类似,H3K4me0紧贴在PHD结合囊中,在域的现有反平行β片上形成额外的β链(图S1). 为了在PHD手指读出H3K4me0的基础上进一步了解分子细节,研究组蛋白识别的动力学事件并定义驱动复合物形成的能量参数至关重要。为此,我们对自由和束缚的AIRE-PHD1进行了多拷贝分子动力学(MD)模拟,以探索基于AIRE-PHD1/H3K4me0相互作用的动力学事件。我们根据构象动力学确定了受肽结合影响最大的残基,并研究了结构域慢集体运动对肽结合的扰动。为了确定H3K4me0组蛋白识别的主要因素,我们接下来使用分子力学/PPoisson-Boltzmann表面积(MM/PBSA)计算分析了天然和突变复合物。MM/PBSA方法是计算给定蛋白质-配体复合物结合自由能的通用工具;该方法将热平均效应与力场/连续溶剂模型相结合,对MD轨迹的一系列代表性快照进行后处理。MM/PBSA已成功应用于计算大量蛋白质-配体相互作用的结合自由能[27]–[30]计算表明,AIRE-PHD1对H3K4me0的识别依赖于静电和极性溶剂化能量项的补偿,主要由非极性相互作用稳定。重要的是,MM/PBSA计算扩展到其他PHD/H3K4me0络合物,显示出能量贡献的类似分布。
结果
H3K4me0肽结合改变AIRE-PHD1动力学
我们使用MD模拟研究了AIRE-PHD1对肽结合的结构和/或动态变化的可能反应。为了部分补偿单个轨迹的不完整MD采样,我们采用了多副本方法,以允许更详尽的构象采样[31]对于每个系统(即自由和束缚AIRE-PHD1),我们进行了五个分子动力学模拟(每个10 ns),其中每个模拟的最后8 ns被连接成一个轨迹并进行分析。AIRE-PHD1的自由初始结构的Cα根-平方(RMSD)偏差(图1A)和装订形式(图1B)相对稳定,RMSD<2.5º。此外,在整个模拟过程中,H3K4me0在现有的AIRE-PHD1反平行β板上形成的额外β链保持不变,并诱导β1链延伸至Glu307残基,这是通过模拟过程中的二级结构分配进行评估的(图1D、E)(我们分别使用单字母代码和三字母代码来识别肽和蛋白质残基)。
图1。游离和结合AIRE-PD1.的构象分析。
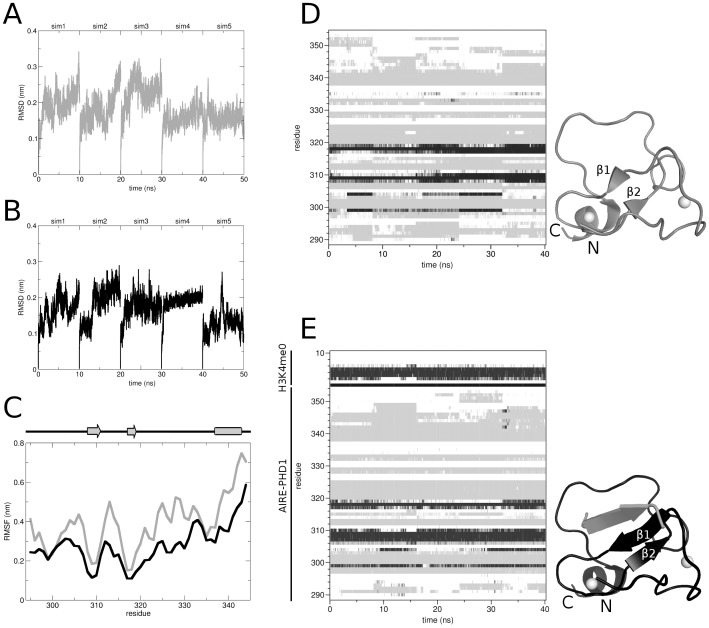
CαRMSD来自(A)自由和(B)束缚的起始AIRE-PHD1结构,作为时间的函数。(C) 自由(灰色)和束缚(黑色)AIRE-PHD1的Cα原子时间平均位置的RMSF。自由(D)和约束(E)AIRE-PHD1的二级结构分配,定义如下do_dssp
[54]作为时间的函数:黑色、白色和灰色分别表示“β-片”、“线圈”和其他二次结构元素。为了进行此分析,将五个MD模拟的最后8 ns连接成一个40 ns的轨迹。H3K4me0与AIRE-PHD1的结合诱导β1链延伸至残基Glu307。自由(灰色)和结合(黑色)AIRE-PHD1的两个代表性结构如图所示:白色球体表示Zn2+离子。
总肽结合限制了AIRE-PHD1探索的构象空间,通过Cα均方根波动(RMSF)的总体降低进行评估(图1C,图S2). 特别是分子间相互作用(总结于表S1)在前9个组蛋白H3残基和结合沟(残基Asn295-Cys310,Ser332-Trp335)之间,AIRE-PHD1在该区域的柔韧性降低。有趣的是,不与肽直接接触的结构域区域,例如连接两条β链(Gly313–Arg316)的环,也降低了它们在复合物中的波动。
接下来,我们想知道H3K4me0肽与AIRE-PHD1的结合是否会影响AIRE-PHD1的协同运动。为此,我们计算了相关矩阵Corrij公司所有Cα原子对。该矩阵描述了动力学过程中所有成对Cα原子围绕其平均位置移动时,它们之间的线性相关性,并提供了相关和反相关运动的全面图片。free相关矩阵的比较(图2A)并绑定AIRE-PHD1(图2B)揭示了两个系统相关运动网络的差异。一方面,自由域几乎没有表现出反相关运动(图2A)另一方面,肽的存在抑制了其中一些运动,并在AIRE-PHD1中诱导了一系列新的相关和反相关运动(图2B)减少短程相关性,增加远端残留物的协同运动。我们详细观察到,在结合后,AIRE-PHD1β链(图2A、C)减少,有利于β1和组蛋白H3肽之间的新相关性(图S3). 此外,肽的结合诱导Glu296和Pro315之间形成相关运动(图2B、D)它们通过复合体中稳定的主干相互作用联系在一起(图S4A). 相反,H3K9和Glu298之间的盐桥形成降低了Glu298-Ala300和Arg303-Glu305之间的相关性,这是由自由域中Arg303和Glu288之间的静电相互作用连接的(图2A、C,图S4B). 最后,在存在配体的情况下,我们还观察到残基Cys322-Pro325和Cys337-Cys340之间存在广泛的反相关(图2B,D)这是由Ser324和Ser338之间的两个氢键快速形成和断裂引起的(图S4C).
图2。自由和束缚AIRE-PHD1的相关运动和主成分分析。
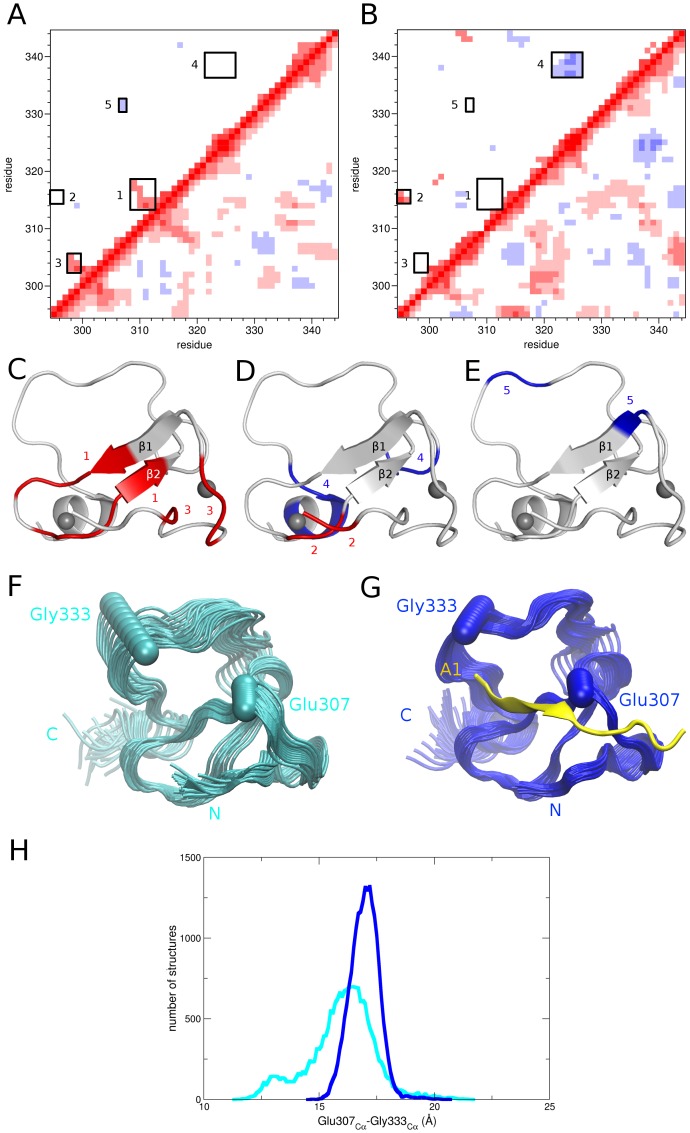
基于残留物(Cα原子)(A)自由和(B)束缚AIRE-PHD1的关联图。原子对之间的相关(正)和反相关(负)运动分别表示为红色和蓝色的颜色梯度。矩阵对角线上方只有|Corrij公司|>报告0.5。文中讨论的相关相关性/反相关性以编号框突出显示,AIRE-PHD1结构上以红色/蓝色报告的(C–E)。自由(F)和束缚(G)AIRE-PHD1的PCA分析。将自由和束缚AIRE-PHD1的Cα运动分别投影到前6和4个特征向量上,得到20个滤波构型的叠加。自由和束缚AIRE-PHD1模拟得到的前6个和4个特征向量分别捕获了总方差累积比例的70%。Glu307和Gly333的Cα原子显示为球体,H3K4me0用黄色表示。(H) Glu307和Gly333的Cα原子之间的距离沿自由(青色)和束缚(蓝色)AIRE-PHD1的动力学分布。
综合起来,RMSF和相关矩阵分析表明,H3K4me0的结合降低了AIRE-PHD1的灵活性,增加了域的协同运动,包括相关运动和反相关运动。
主成分分析
为了表征整个领域的运动,我们对轨迹产生的协方差矩阵进行了主成分分析(PCA)[32]PCA可以将相关变量的原始空间转换为独立变量的缩减空间(即主成分或特征向量)。主成分分析识别残基组的相关低能位移,并通过将轨迹投影到降维空间来强调主要蛋白质运动的幅度和方向,从而提取轨迹中捕获的慢模式[31]利用这种方法,我们确定了参与最相关的集体构象变化的蛋白质区域,并阐明了配体结合诱导的AIRE-PHD1动态调制。首先,我们观察到,与AIRE-PHD1/H3K4me0复合体相比,自由AIRE-PHD1的前几个特征向量捕获的累积方差始终较低。特别是,在自由约束AIRE-PD1中,总方差的累积比例的70%由前六个和四个特征向量捕获(表S2). 这与肽结合增加AIRE-PHD1相关运动的事实相符(图2B). 通过计算主成分的平方根内积(RMSIP),对模拟结果进行比较[33]揭示了自由和束缚AIRE-PHD1的基本运动特征不同(RMSIP=0.52),并描述了不同的基本子空间。沿着这些组分的MD轨迹投影表明,自由AIRE-PHD1的基本运动主要是由残基Arg328-Thr334组成的环向β1链的运动(图2B,E). 值得注意的是,这个内在领域“呼吸”(图2F)在H3K4me0肽的存在下被强烈还原,该肽在“开放”构象中阻断了结构域(图2G). 在整个模拟过程中,我们始终观察到Glu307和Gly333的Cα原子之间的采样距离分布变窄(图2H).
天然和突变AIRE-PHD1/H3K4me0配合物的MM/PBSA计算
接下来,我们研究了驱动AIRE-PHD1和组蛋白H3肽之间天然和突变形式相互作用的能量参数,并将计算结果与可用的实验数据进行了比较。为此,我们利用MD模拟生成的大量构象数据,使用MM/PBSA方法对野生型复合体进行结合自由能计算。该方法表示结合自由能(ΔGcomp公司)作为络合物的自由能与受体加配体的自由能之差,在多个轨迹快照上的平均值[34]接下来,我们在同一组快照上进行丙氨酸突变,并重新计算相关的结合自由能差异(ΔΔGcomp公司).结果然后与ITC测量的可用实验热力学数据进行比较[15],[20]。使用内部脚本进行计算(请参见材料和方法)这提供了程序的快速自动化。
值得注意的是,MM/PBSA方法允许快速估计结合自由能的变化,但通常不会复制绝对结合自由能值。然而,它通常表现出与实验的良好相关性,从而代表了计算和比较结合自由能变化的效率和功效之间的公平折衷。事实上,在我们的计算中,我们观察到计算出的结合自由能比实验得到的大(表1)这是一种在其他系统中已经观察到的高估。这种行为通常被归因于忽略了熵贡献,这是这些计算中的典型近似值[27],[35]–[39]在这里,我们主要关注野生型和突变复合物之间的差异,尽管该方法存在固有的局限性,但我们观察到计算和实验之间具有良好的定性一致性,相关系数为第页0.85(图3).
表1。野生型和突变型AIRE-PHD1/H3K4me0复合物的MM/PBSA结合自由能(kJ/mol)。
|
突变体 |
实验 |
计算 |
极性贡献 |
非极性贡献 |
|
1ΔG经验
|
2ΔΔG经验
|
三ΔGcomp公司
|
4ΔΔGcomp公司
|
5ΔG库尔
|
6ΔG秒
|
7ΔG极地的
|
8ΔG虚拟数据仓库
|
9ΔG非营利组织
|
10ΔG非极性的
|
|
野生型
|
−29.73 |
0 |
−170.7(4.0) |
0 |
−4337.7(16.3) |
4416.4(15.6) |
78.7 |
−217.6(2.7) |
−31.8(0.2) |
−249.4 |
|
谷氨酸307Ala
|
−24.70 |
5 |
−164.5(3.6) |
6.1 |
−3779.3(16.4) |
3860.0(16.5) |
80.6 |
−214.2(2.8) |
−30.9(0.2) |
−245.1 |
|
谷氨酸298Ala
|
−24.70 |
5 |
−147.7(3.8) |
22.9 |
−3632.3(16.4) |
3742.5(15.1) |
110.2 |
−226.6(2.6) |
−31.3(0.2) |
−257.9 |
|
R8A型
|
−24.28 |
5.5 |
−136.1(3.4) |
34.5 |
−3384.0(13.4) |
3486.6(12.7) |
102.6 |
−209.3(2.5) |
−29.5(0.1) |
−238.7 |
|
天冬氨酸304Ala
|
−21.77 |
8 |
−117.2(3.1) |
53.4 |
−3681.2(16.8) |
3817.2(16.0) |
136 |
−222.1(2.6) |
−31.2(0.2) |
−253.2 |
|
阿斯普297Ala
|
−21.72 |
8 |
−132.7(4.1) |
38 |
−3507.5(16.0) |
3634.3(15.2) |
126.8 |
−228.0(2.5) |
−31.5(0.2) |
−259.5 |
图3。天然和突变AIRE-PHD1/H3K4me0配合物的MM/PBSA计算。
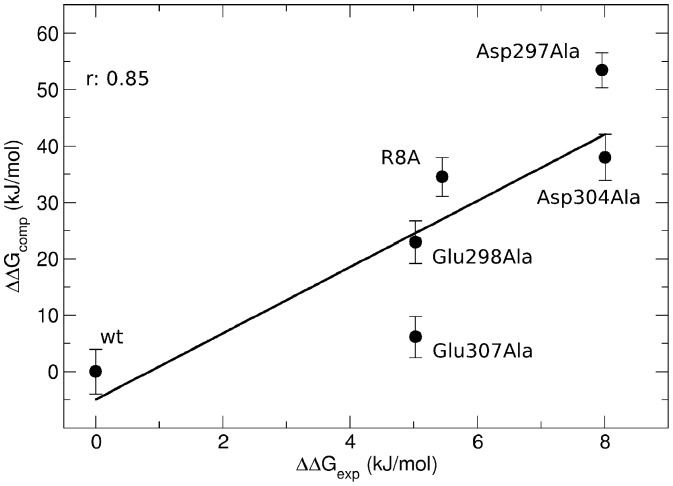
实验结合自由能差曲线(ΔΔG经验)与计算的结合自由能差(ΔΔGcomp公司)AIRE-PD1/H3K4me0丙氨酸突变体的表达。
将结合自由能分解为其组分,包括范德华、静电、极性溶剂化和非极性溶剂化互作用能项,确定了野生型和突变体结合亲和力的主导因素。一方面,库仑(ΔG库尔)范德瓦尔斯(ΔG虚拟数据仓库)和非极性溶剂化项(ΔG非营利组织)有利于复合物的形成。另一方面,正极性溶剂化作用(ΔG秒)由于肽和AIRE-PHD1的极性残基和带电残基的去溶能不利。总的来说,这种高脱溶能量损失不能被库仑相互作用完全补偿,从而导致极性项对结合自由能变化(ΔG极地的>0). 相反,非极性能量项ΔG非极性的由范德瓦尔斯项和非极性溶剂化项之和(远小于范德瓦尔项)形成的,促进了络合物的形成(ΔG非极性的<0). 综上所述,这些数据表明复合物由非极性相互作用稳定,并由极性贡献调节(表1). 总的来说,丙氨酸突变降低了库仑溶剂化和极性溶剂化对野生型复合物的贡献,导致正极性项的总体增加,从而对抗结合。始终ΔG极地的与实验结果有很好的相关性(第页 = 0.83),而ΔG非极性的没有显示出实质性的变化。
识别H3K4me0的其他PHD手指的MM/PBSA计算
受AIRE-PHD1/H3K4me0复合物令人鼓舞的结果的启发,我们接下来想知道MM/PBSA方法是否也可以用于计算和排序H3K4me0肽复合物中其他PHD指的结合ΔG,包括CHD4-PHD2、BHC80-PHDD、TRIM24-PHD和BRPF2-PHD1(表2). 五个10 ns长MD模拟的最后一纳秒被连接成一个单一轨迹(5 ns),用于MM/PBSA计算。尽管使用了不同的实验条件(主要差异在于缓冲液类型和离子强度,而pH值和温度是可比较的)和技术(荧光和ITC)来确定离解常数(表2和表S3),我们观察到结合的实验和计算ΔΔG之间有很好的相关性(第页 = 0.96) (图4A). 重要的是,在所有五个配合物中,H3K4me0的识别通过非极性项(ΔG)稳定非极性的<0),而ΔG极地的由负静电和正极性溶剂化组分组成的术语对抗结合。值得注意的是,ΔG极地的与ΔΔG相关良好经验(第页 = 0.88),而ΔG非极性的不同复合体之间没有显著变化(表2).
表2。PHD指/H3K4me0配合物的MM/PBSA结合自由能(kJ/mol)。
|
|
含H3K4me0的络合物 |
实验 |
计算 |
极性贡献 |
非极性贡献 |
|
1ΔG经验
|
2ΔΔG经验
|
技术和实验条件 |
三ΔGcomp公司
|
4ΔΔGcomp公司
|
5ΔG库尔
|
6ΔG秒
|
7ΔG极地的
|
8ΔG虚拟数据仓库
|
9ΔG净现值
|
10ΔG非极性
|
|
AIRE公司
|
−29.73 |
0 |
ITC、20 mM磷酸盐缓冲液、150 mM氯化钠、2 mM 2-巯基乙醇、50 mM氯化锌2pH值7.2 |
−170.1(3.9) |
0 |
−4337.7(16.3) |
4417.0(15.7) |
79.3 |
−217.6(2.7) |
−31.8(0.2) |
−249.4 |
|
TRIM24阀内件
|
−28.90 |
0.83 |
ITC、20 mM磷酸盐缓冲液、150 mM氯化钠、2 mM 2-巯基乙醇、50 mM氯化锌2pH值7.2 |
−167.8(3.9) |
2.3 |
−4506.5(20.9) |
4562.8(18.4) |
56.3 |
−195.0(2.4) |
−29.2(0.1) |
−224.2 |
|
瑞士法郎4
|
−27.10 |
2.63 |
色氨酸荧光,20 mM磷酸钠,150 mM NaCl,10 mM DTT,1 mM NaN3,pH 7.2 |
−137.0(3.8) |
33.1 |
−4035.3(27.6) |
4156.0(23.7) |
120.7 |
−226.2(3.7) |
−31.5(0.1) |
−257.6 |
|
BHC80型
|
−25.57 |
4.16 |
ITC,25 mM三氯化氢,50 mM氯化钠,2 mM 2-巯基乙醇,pH 7.2 |
−127.8(4.0) |
42.3 |
−2245.8(11.0) |
2355.5(10.6) |
109.7 |
−208.8(3.9) |
−28.7(0.1) |
−237.5 |
|
BRPF2型
|
−20.85 |
8.88 |
ITC,50 mM三氯化三钠,100 mM氯化钠,pH 7.5 |
−104.1(4.9) |
66 |
−4582.6(34.7) |
4732.8(32.4) |
150.2 |
−221.0(3.7) |
−33.4(0.2) |
−254.3 |
图4。识别H3K4me0的PHD手指的MM/PBSA计算。
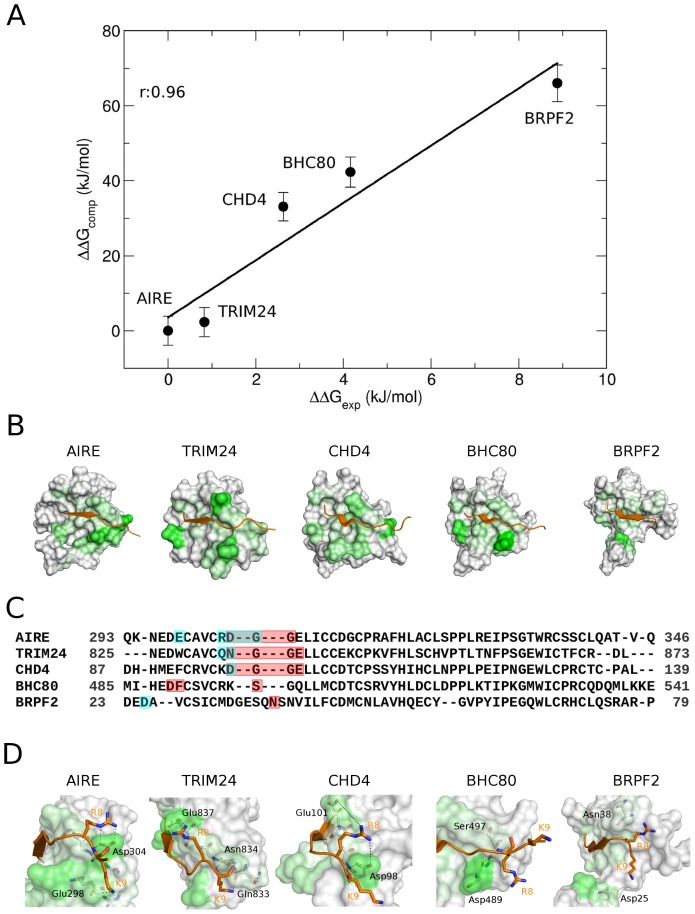
(A) 实验结合自由能(ΔG)之间的相关性经验)和计算的结合自由能(ΔΔGcomp公司)H3K4me0-绑定PHD手指。(B) 与肽结构域分子间接触相关的能量贡献(库仑能和范德华能)的表示。为了清楚起见,归一化相互作用能量仅映射在PHD手指表面上,范围从白色(无贡献)到绿色(高贡献)。H3K4me0表示为橙色丝带。(C) MultiSeq生成的H3K4me0-绑定PHD手指的结构对齐[55]与H3R8和H3K9相互作用的残留物分别以红色和青色突出显示;与H3R8和H3K9相互作用的残基以灰色突出显示。(D) H3R8和H3K9(以棒状显示)与PHD指状残基之间分子间相互作用的表示;虚线表示极性触点。
分子间接触分析
利用对五种不同配合物进行的MD模拟,我们对与肽域分子间接触相关的能量贡献(库仑和范德瓦尔斯能量)进行了比较分析。这些接触是根据它们的分子间距离(<3º)和它们在MD模拟期间的统计发生率(>30%)以及它们的相关能量来定义的(表S1)映射到域表面(图4B). 有趣的是,我们观察到组蛋白肽的前6个残基与等效残基建立了类似的接触,如基于结构的比对所定义的(图4C). 值得注意的是,主要差异涉及残基H3R8和H3K9(H3A7的贡献可以忽略不计),这表明接触次数较高(表S1)和有利的相互作用能(图4B、D;表S1)在那些PHD手指中显示出对H3K4me0的最佳亲和力。相反,缺乏这些相互作用的PHD手指,如BRPF2-PHD1,表现出最低的亲和力。总的来说,这些结果表明对H3R8和H3K9的识别可能会调节相互作用的精细细节,从而赋予不同PHD域的阅读特异性。
讨论
PHD手指的结构和功能在过去几年中受到了深入的研究,形成了30多个复杂结构,为其与组蛋白肽的相互作用提供了“静态描述”[7],[10],[40]令人惊讶的是,到目前为止,很少有人致力于这些络合物的动力学表征以及驱动络合物形成的能量参数的定义/预测。在这种情况下,计算方法提供了直接观察结合事件和剖析决定结构域肽识别的动态和能量决定因素的机会。作为PHD手指对组蛋白H3识别的动态和能量理解的第一步,我们首先关注AIRE-PHD1与H3K4me0复合物的动力学和热力学研究。AIRE-PHD1是非修饰组蛋白H3读取器的典型例子。然后将热力学结果与在属于同一类H3K4me0读卡器的其他PHD指上进行的计算进行比较。
对AIRE-PHD1进行的多拷贝MD模拟表明,与H3K4me0肽的结合总体上降低了AIRE-PHD1的灵活性。在与H3A1和H3K4直接接触的特定区域,包括N末端残基和位于回路Ser332-Trp335上的残基,显示RMSF波动减少(图1C,图S1). 这个观察结果与之前的完全一致15N弛豫研究表明,相同的区域在底物识别后,在纳秒时间尺度上变得更加坚硬,这是通过在肽存在下异核NOE值增加来评估的[15]分析自由和结合AIRE-PHD1的相关图表明,H3K4me0肽的结合减弱了一些短程关联,并诱导了新的长程关联,涉及与肽直接接触的残基和作用表面远端的残基,从而桥接了一些原本遥远的残基。
为了检查AIRE-PHD1中的协同运动,并评估这些运动是如何受到肽结合的影响,我们使用PCA。特别是,对基本结构域运动的分析强调了自由AIRE-PHD1中存在的“扑动”运动,涉及由残基Arg328-Thr334和β1的N末端边缘组成的环。重要的是,这个可能与结构域功能相关的固有结构域“呼吸”在H3K4me0结合时被阻断在开放构象中。总的来说,肽识别强烈影响了动态相关氨基酸的结构域网络,改变了结构域结构。我们假设这种动态事件可能发生在全长AIRE的背景下,并可能在AIRE染色质相互作用的框架内具有功能相关性。剩下的一个挑战是确定该网络的扰动在多大程度上有助于调节结构域功能和/或其与组蛋白H3尾部的相互作用。
PHD手指活动功能障碍与许多疾病之间的联系表明这些系统具有强大的治疗潜力[7]在此,AIRE-PHD1为PHD指和非甲基化组蛋白尾之间相互作用的计算模型的生成提供了一个有用的基准,因为在极为可控的条件下(根据pH、温度和离子强度),可以获得关于天然和突变复合物的数个实验热力学数据[15],[20]利用实验信息和从MD模拟中获得的丰富构象数据,我们采用MM/PBSA方法来计算与AIRE-PHD1/H3K4me0复合物的天然和丙氨酸突变体相关的结合自由能。计算的结合自由能与实验数据有很好的相关性(第页 = 0.85),表明该方法用于估算结合自由能是可靠的生物信息学对于天然和突变络合物,从溶剂可及表面积(SASA)和范德瓦尔斯项之和获得的非极性能量值有助于络合物的结合自由能。另一方面,极性能量项显著拮抗肽与天然和突变AIRE-PHD1的结合。这种影响主要是由于极性残基去溶剂化项的成本很高,相对于有利的库仑相互作用能来说,这更高,这意味着AIRE-PHD1和H3K4me0肽之间的分子间静电相互作用不足以完全支付去溶剂化惩罚。然而,不利的ΔG极地的项主要由负ΔG补偿非极性的导致非常有利的结合自由能的术语。所以复合物的形成由非极性相互作用稳定,并由极性相互影响调节。这种热力学特征已经在JMJD2A的Tudor结构域与H3/H4组蛋白尾部的复合物中观察到[41]这可能是表观遗传读者识别组蛋白尾部机制中的一个共同主题。正如预期的那样,丙氨酸突变减少了静电对结合自由能的贡献,并减少了极性溶剂化项,导致不利极性项ΔG的总体增加极地的拮抗结合;非极性贡献主要不受丙氨酸突变的影响。基于这些结果,我们假设未来MM/PBSA可能代表一种稳健的预测方法生物信息学其他突变和表观遗传修饰对AIRE解码组蛋白H3能力的影响。特别是,在专注于AIRE在转录激活中的病理生理作用的分子研究背景下,MM/PBSA计算可能会成为一种快速工具,帮助实验人员合理设计组蛋白H3结合活性改变的AIRE突变体,以调节其转录活性。
一旦我们证明MM/PBSA可以构成一种可靠的计算方法来计算我们模型系统的结合自由能,我们接下来想知道该工具是否可以普遍应用于识别H3K4me0的其他PHD手指的能量分析。值得注意的是,我们观察到计算的结合自由能和实验的结合自由能量之间有很好的相关性(第页 = 0.96),因此在所有PHD手指中,H3K4me0的识别通过非极性项(ΔG)稳定非极性的<0),而ΔG极地的不利约束项(ΔG极地的>0). 特别是ΔG极地的与实验结果有很好的相关性(第页 = 0.88)表明PHD手指静电特性的差异可能会影响结合亲和力。为了进一步研究这一方面,我们分析了五种配合物的MD模拟,详细研究了界面残基子集的相互作用能量。该分析指出,H3K4me0的前6个残基与不同PHD指中的等效残基建立了类似的接触。重要的是,与亲和力较低的PHD指状物(例如BRPF2-PHD1)相比,那些对H3K4me0具有较高亲和力的配合物(例如AIRE-PHD1)与残基H3R8和H3K9表现出较高的相互作用。这些观察结果表明,H3R8和H3K9可能参与PHD手指识别的微调,从而确定这类H3K4me0读取器的选择性。通过计算和实验评估,H3R8丙氨酸突变降低了AIRE-PHD1的H3亲和力,这一观察结果支持了这一假设。总的来说,这些数据对于MD结合MM/PBSA的应用是非常令人鼓舞的,它是一种有价值的工具,可以快速分析支配这类表观遗传效应器组蛋白解码的能量决定因素。
正如AIRE-PHD1已经证明的那样,可以想象MM/PBSA也可以应用于组蛋白阅读器的这一子集,以预测生物信息学丙氨酸突变的影响,并表征该亚类表观遗传学读者的单个成员对组蛋白H3识别的精细热力学细节。PHD手指确实是作为可药物化的蛋白质相互作用域类别出现的,它们代表了药物发现的新前沿,对未来治疗学的发展具有巨大潜力[42]建立了这类PHD手指不同成员对组蛋白识别的共同原理和差异,这项工作为进一步研究其他H3K4me0读取器铺平了道路,并可能有助于药物设计研究,重点关注PHD小抑制剂的开发。未来将有兴趣测试该方法的稳健性,以验证这组复合物是否可以用作训练集来预测同类其他PHD手指(例如Sp140、NSD1、Sp110)的亲和力排名。未来的研究还将致力于探索这些计算方法在PHD手指能量分析中的应用,以识别其他表观遗传特征,如H3K4me3、H3K4me9或乙酰化标记,因为有几种高分辨率的复合物结构可用。
材料和方法
AIRE-HD1 MD模拟
自由形式的AIRE-PHD1结构(1xwh)和结合到H3K4me0肽(残基1-10)(2ke1)的结构用于MD模拟。使用Gromacs 4.0.7软件包进行MD模拟[43]液体模拟(OPLS)力场的优化参数[44].通过添加适当数量的钠反离子来中和系统。执行能量最小化程序和200 ps的位置约束阶段,以缓解不利的相互作用;随后在NVT系综中进行300 ps的MD模拟,以平衡系统(T=296 K)。生产运行在NPT装置中进行,P=1 bar,T=296 K,持续10 ns,时间步长为2 fs。用粒子网格Ewald(PME)方法处理长程静电,使用1.1 nm截止。对于Lennard-Jones相互作用,使用1.1纳米截止。每10个MD步骤更新一次配对列表。进行了五个独立的10 ns长MD模拟(50 ns生产运行),以便进行更好的构象取样,并对结合自由能进行统计验证。五个独立模拟的起始结构是从沉积的核磁共振束中提取的。
通过计算平均结构的RMSD值和MM/PBSAΔG的累积平均值来评估结构和能量收敛绑定值分别作为时间的函数(图S5A、B).
为了进一步验证收敛性,我们还将每个副本的模拟长度扩展到50 ns。50 ns后,所有模拟均具有稳定的RMSD曲线(图S6). 由于10 ns和50 ns模拟所探索的构象空间是相同的(请参见下文),因此对10 ns长的模拟进行了构象分析。
H3K4me0肽复合物中其他PHD手指的MD模拟
如上所述,对CHD4-PHD2(2l75)、BHC80-PHDD(2puy)、TRIM24-PHD(3o37)和BRPF2-PHD1(2I43)与H3K4me0肽的复合物进行MD模拟。对于与H3肽复合物中的CHD4-PHD2,在进行MD模拟之前从H3K9中去除甲基。对于TRIM24-PHD,计算中仅使用了残基Asn825-Pro885,这是基于溴代多巴胺不参与PHD-H3肽相互作用的结构观察[18]对于BRPF2-PHD1,去除残基Gly13-Ser18,生成两条分离链,包括PHD指和12个残基长度的组蛋白尾(结构是通过融合构建物确定的,既包含PHD指又包含组蛋白尾,而结合亲和力是通过用相应的组蛋白肽滴定BRPF2-PHD1来确定的[19]). 对每个综合体进行五次独立的10 ns长MD模拟(50 ns生产运行)。对于NMR测定的配合物,从沉积的结构系综(2l75,2l43)中提取了五个起始结构,而对于晶体结构(2puy,3o37),五个独立的动力学运行使用了不同的种子数。根据RMSD和MM/PBSAΔG的平均结构计算累积平均值,评估结构和能量收敛绑定值(图S5B,A).
主成分分析
协方差矩阵的主成分分析(PCA)确定沿分子动力学模拟生成的轨迹的主要低频大尺度运动。这种统计方法用于描述最相关的运动,使用一个新的基集直接反映系统所经历的集体运动。该方法可以过滤决定系统运动的主要模式的噪声,从而降低MD轨迹的维数[32]PCA需要构造协方差矩阵覆盖(cov)基于给定原子集相对于系综平均位置的三维位置起伏,通过最小二乘拟合去除整体旋转和平移运动。元素秒ij公司矩阵的冠状病毒是(方程式1):
哪里第页我和第页j个是向量的Cα我和Cαj个轨迹中的位置;<r我>t吨和<rj个>t吨是第页我和第页j个时间平均值(t吨)超过MD轨迹;指数i和j表示氨基酸残基。数据通常由根据相关矩阵计算的相关图表示,其元素由
该图允许识别具有相关(在同一方向)和反相关(在相反方向)运动的残差对/组。协方差矩阵c的对角化ov型提供了一组正交的特征向量,每个特征向量由一个特征值定义,分别表示运动的方向和幅度,其中第一个特征向量表示对系统总波动的最大贡献,第二个特征向量是第二个最大贡献,依此类推。对AIRE-PHD1(残基Asn295-Thr344)结构系综的Cα坐标进行PCA计算,该结构系综由游离AIRE-PHD1的五个MD的最后8ns的串联产生,并分别与H3K4me0肽复合。
本质子空间与收敛性评估的比较
为了比较自由和束缚AIRE-PHD1模拟中识别的特征向量所描述的基本子空间,我们通过计算两组相应特征向量之间的均方根内积(RMSIP)来计算两个子空间之间的重叠:
其中v我和wj个是我-th和j个-两个集合的第个特征向量。RMSIP的范围从零(正交、非重叠子空间)到一(即相同子空间)。RMSIP通常是在定义基本子空间的十个特征值最大的特征向量上计算的[33].
RMSIP的计算还可以用于比较描述相同仿真的不同时间窗口的特征向量,以验证仿真收敛性[45].
因此,为了评估模拟收敛性,我们将单个模拟分为三个不同的时间窗口(2-5 ns;2-8 ns;2-10 ns),并计算了它们相应的特征向量。接下来,我们计算RMSIP以验证这些特征向量所描述的单个子空间是否相似。重要的是,所有RMSIP值均大于0.85,表明这些特征向量所描述的子空间之间存在较大重叠(表S4). 重要的是,10 ns和50 ns仿真所描述的子空间基本相同,正如两个仿真的前10个特征向量之间的高RMSIP值所示(表S5). 我们的结论是,在10毫微秒时,模拟已经达到收敛,并且2到10毫微斯的生产运行为这些系统的有意义分析提供了足够的采样。
基于MD的结合自由能计算
根据MM/PBSA方法测定结合自由能的方法已在前面描述[34]蛋白质分子与溶液中配体分子的结合自由能定义为:
进行MD模拟以生成结构的热力学加权集合(在我们的示例中,是时间等距快照的集合)。自由能项计算为所考虑结构的平均值:
能量项EMM(毫米)定义为:
其中E整数表示键能、角能和扭角能,以及E库尔和ELJ公司分别表示分子内静电和范德瓦尔斯能量。
溶剂化项G解决在里面等式(7)被分成极性G极地的和非极性贡献,G非极性的
[34]:
在本工程中G极地的和G非极性的使用APBS(自适应泊松-玻耳兹曼解算器程序)进行计算[46].极性贡献G极地的指将溶质从低介电常数(ε=1)的连续介质转移到介电常数为水的连续介质(ε=80)所需的能量。G公司极地的使用非线性泊松-玻尔兹曼方程进行计算。栅格间距自动设置为上限0.5º。温度设置为296 K,盐浓度为0.15 M。非极性贡献G非极性的被认为与溶剂可及表面积(SASA)成正比:
哪里γ = 0.0227千焦摩尔−1Å−2和β = 0千焦摩尔−1
[47]使用半径为1.4º的探针确定介电边界。
基于MM/PBSA方法的结合自由能计算可以根据三轨道法(TTM)或单轨道法(STM)进行。TTM需要对三种系统成分(复合物、游离配体和游离受体)进行三次单独的MD模拟。这是一种需要计算的方法,容易产生结构噪声[28],[30]相反,STM需要复合物的单一轨迹运行,从而直接从复合物结构中提取蛋白质和配体结构[28]从而使E归零整数术语。在这种情况下,假设蛋白质和配体在结合形式和自由形式中的行为相似。这一假设对于PHD指状物是合理的,因为它们在结合时不会发生结构重排[6]–[8]如果肽结构重排发生在与PHD指结合时。在所研究的系统中,肽与PHD指结合时总是采用β链构象。因此,可以合理假设不同复合体中的熵项非常相似,并且在计算ΔΔG时会合理抵消comp公司使用AIRE-PHD1/H3K4me0作为参考值(ΔΔG)计算差异comp公司 = ΔGAIRE公司-ΔG博士). 因此,根据STM协议进行了MM/PBSA计算。
在MM/PBSA近似值内<EMM(毫米)>+<G解决>解释了与络合物形成相关的焓变化。计算确定结合自由能需要计算熵对复杂形成的贡献,包括溶质转动、平移和振动自由度的构象变化。溶质的熵贡献通常通过准手征方法(例如Schlitter方程)或正规模式分析进行估算[48]熵计算需要对自由能源景观进行全面采样,这是一个计算要求极高的步骤,可能会导致不可靠的结果[36]标准误差通常比与其他能量项相关的误差大一个数量级[49]此外,正常模式分析估计通常是非常定性的[50]在短动态时间范围内的构型熵估计可能不重要[51]基于这些考虑,以及我们主要对ΔΔG感兴趣的事实comp公司(详见下文)我们决定在计算中忽略熵项。MM/PBSA计算考虑了五个MD模拟(即125个等时距帧)最后纳秒的平均值。标准误差(SE)计算如下:
其中σ是标准偏差,N是计算中使用的结构数量(125)。
丙氨酸突变
在野生型复合物丙氨酸突变后没有重大构象变化的情况下,可以使用MM/PBSA方法对野生型模拟中的快照进行丙氨酸突变复合物的结合自由能计算,而不是对单个突变复合物进行模拟[29],[34]该方案已成功应用于研究各种蛋白质相互作用[28]–[30]简单地说,对MD模拟获得的野生型结构进行后处理,以引入丙氨酸突变。因此,计算结合自由能值可以表示为野生型和突变复合物之间的结合自由能差异(ΔΔGcomp公司). 在这些计算中,忽略了熵,假设相似配体结合自由能的熵贡献在相对比较中抵消了[28],[30],[34],[37],[52].
GMXAPBS工具
尽管免费提供的软件Gromacs 4.0.7很受欢迎[43]和APBS[46]为了直接使用MD输出作为结合自由能计算的输入,自动组合这两个程序是不可用的。为了方便两个程序之间的接口,我们编写了一系列Bash/Perl脚本,直接对MD模拟生成的结构执行MM/PBSA计算。运行脚本只需要三个MD仿真文件:轨迹文件(TRR或XTC)、拓扑文件(TPR)和索引文件(NDX)。对于自定义的力场,该工具还需要拓扑和参数文件。计算以自动方式组织,可以在PBS队列系统中并行运行。图S7总结了脚本工作流的示意图表示。简而言之,Gromacs MD模拟生成的结构是作为PDB文件从轨迹中提取的。接下来,结构进行能量最小化(长度可由用户确定),在此期间,范德瓦尔斯项以双精度计算。然后,PDB文件被转换为PQR文件编辑confGromacs工具。APBS通过APBS辅助程序计算溶剂化(极性和非极性)和静电项库仑泊松-玻尔兹曼方程需要生成一个具有合适网格的方框。为此,蛋白质在每个维度的极端坐标都会自动从结构文件中提取出来。将20和10Ω添加到每个值中,以分别设置粗网格和细网格的限制。然后,我们的工具自动计算网格小于0.5º时适用于APBS计算的网格点数量。最后,当所有计算完成后,将所有结合能项相加,以获得沿轨道的平均结合自由能。在我们的集群(HP 300核、2.9 GHz、4 Gb RAM、InfiniBand连接、Maui/TORQUE队列系统)中,每天已经进行了3000个结构的计算。GMXAPBS工具也被改编为执行丙氨酸突变的后处理协议:GMXAPBS脚本可以将任何氨基酸突变为丙氨酸,将蛋白质的残基截断到Cβ原子,并添加缺失的氢原子以完成C的四面体配位β原子。随后,进行MM/PBSA计算,以计算丙氨酸突变对结合亲和力的能量影响。GMXAPBS是全自动的,很容易适应任何蛋白质和蛋白质结合系统。脚本被广泛注释以便于定制,输出是报告计算术语的文本或pdf文件。该工具可根据要求提供。
支持信息
图S1
AIRE-PHD1/H3K4me0复合体的表面图。AIRE-PHD1(白色卡通和表面)和H3K4me0(橙色卡通)的综合体。与H3A1、H3R2、H3K4、H3R8和H3K9相互作用的AIRE-PHD1残基分别显示为绿色、品红色、青色、粉红色和紫色。虚线表示复合物的极性触点和Zn的选择2+离子用灰色球体表示。
(畅通节能法)
图S2
自由(青色)和束缚(蓝色)AIRE-PHD1的五个复制品的Cα原子从其时间平均位置的RMSF。
(畅通节能法)
图S3
AIRE-PHD1(残基295-344)和H3K4me0(残基1-5,黑线)的基于残基(Cα原子)的关联图。箭头表示AIRE-HD1β1链与组蛋白尾部形成的额外β链之间的相关性。
(畅通节能法)
图S4
与中方框1、2和3所述相关性对应的相互作用
图2
.左侧显示了沿自由(青色)和束缚(蓝色)AIRE-PHD1动力学的特定距离分布,右侧显示了自由(青蓝色)和绑定(蓝色)空气-PHD1的两个代表性结构,灰色球体和黄色卡通表示Zn2+离子和组蛋白尾。(A) Glu296和Pro315主链原子之间的相互作用,(B)Glu298和Arg303侧链之间的盐桥,(C)Ser324和Ser338之间的氢键。
(畅通节能法)
图S5(A) Cα原子相对于不同系统模拟所得平均结构的RMSD值的累积平均值。(B) 不同系统五次模拟所得MM/PBSA值的累计平均值。
(畅通节能法)
图S6
CαRMSD来自(A)自由和(B)束缚启动AIRE-PHD1结构,作为50 ns轨迹中时间的函数。
(畅通节能法)
图S7
GMXAPBS工作流程示意图。
(畅通节能法)
表S1
MD模拟期间PHD手指和H3K4me0之间的原子间接触以及相关的相互作用能量。对每条轨迹的最后8纳秒进行分析。在该分析中,“接触”定义了在超过30%的总模拟帧中发生的任何一对原子之间的相互作用距离(<3º)。等效残留物,如图1所示的结构线形所定义图4C,与相应的能量贡献(kJ/mol)在同一条线上报告。
(文件)
表S2
自由和束缚AIRE-PHD1动力学的前六个特征向量捕获的总方差的方差比例和累积比例。
(文件)
表S3
用于MM/PBSA计算的PHD-H3K4me0络合物概要。
(文件)
表S4
自由和束缚AIRE-PHD1轨迹(#)的三个不同时间窗口(2-5、2-8和2-10 ns)获得的特征向量之间的RMSIP值。
(文件)
表S5
从自由和有界AIRE-PD1轨迹的三个不同时间窗口(2-10、2-30和2-50 ns)获得的特征向量之间的RMSIP值(#)。
(文件)
致谢
我们感谢Telethon基金会和Cariplo基金会的财政支持。
我们感谢Michela Ghitti博士、Giacomo Quilici和Mike P.Williamson教授对手稿的批判性阅读。
资金筹措表
作者得到了Telethon基金会的资助。DS拥有由“维塔Salute-Milan大学”支持的博士生奖学金。资助者在研究设计、数据收集和分析、决定出版或编写手稿方面没有任何作用。
工具书类
-
1Bannister AJ,Kouzarides T(2011)组蛋白修饰对染色质的调节。单元格Res21 (3): 381–395.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
2.Chi P,Allis CD,Wang GG(2010)共价组蛋白修饰——人类癌症中的误写、误解和误读。Nat Rev癌症10 (7): 457–469.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
三。Taverna SD,Li H,Rutenburg AJ,Allis CD,Patel DJ(2007)染色质结合模块如何解释组蛋白修饰:来自专业拾荒者的教训。自然结构分子生物学14 (11): 1025–1040.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
4Khorasanizadeh S(2011)《甲基化组蛋白的识别:新的曲折和变化》。当前操作结构生物21 (6): 744–749.[内政部] [公共医学] [谷歌学者]
-
5Yun M,Wu J,Workman JL,Li B(2011)组蛋白修饰的读者。单元格Res21 (4): 564–578.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
6Musselman CA,Kutateladze TG(2011)用PHD手指手工挑选表观遗传标记。核酸研究219: 9061–9071.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
7Musselman CA,Kutateladze TG(2009)PHD手指:表观效应器和潜在药物靶点。分子间相互作用9 (6): 314–323.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
8Bienz M(2006)PHD指,核蛋白相互作用域。生物化学科学趋势31 (1): 35–40.[内政部] [公共医学] [谷歌学者]
-
9Baker LA、Allis CD、Wang GG(2008)《人类疾病中的PHD手指:由误解表观遗传标记引起的疾病》。突变研究647 (1–2): 3–12.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
10Sanchez R,Zhou MM(2011)《博士手指:一个多才多艺的表观基因组读取器》。生物化学科学趋势36 (7): 364–372.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
11Li H,Ilin S,Wang W,Duncan EM,Wysocka J,et al.(2006)NURF的BPTF PHD指定点读取组蛋白H3K4me3的分子基础。自然442 (7098): 91–95.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
12Shi X,Hong T,Walter KL,Ewalt M,Michishita E,et al.(2006)ING2 PHD结构域将组蛋白H3赖氨酸4甲基化与活性基因阻遏联系起来。自然442 (7098): 96–99.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
13Palacios A、Munoz IG、Pantoja-Uceda D、Marcaida MJ、Torres D等(2008)ING4识别组蛋白H3K4me3的分子基础。生物化学杂志283 (23): 15956–15964.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
14Lan F、Collins RE、De Cegli R、Alpatov R、Horton JR等(2007年)。未甲基化组蛋白H3赖氨酸4的识别将BHC80与LSD1介导的基因抑制联系起来。自然448 (7154): 718–722.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
15Chignola F,Gaetani M,Rebane A,Org T,Mollica L,et al.(2009)与非修饰组蛋白H3尾部复合物中自身免疫调节物第一个PHD指的溶液结构揭示了H3R2甲基化的拮抗作用。核酸研究37 (9): 2951–2961.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
16Chakravarty S,Zeng L,Zhou MM(2009)人类自身免疫调节物PHD指对组蛋白H3的结构和位点特异性识别。结构17 (5): 670–679.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
17Mansfield RE、Musselman CA、Kwan AH、Oliver SS、Garske AL等人(2011)CHD4的植物同源结构域(PHD)指是组蛋白H3结合模块,优选未修饰的H3K4和甲基化的H3K9。生物化学杂志286 (13): 11779–11791.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
18Tsai WW,Wang Z,Yiu TT,Akdemir KC,Xia W,et al.(2010)TRIM24将非标准组蛋白特征与乳腺癌联系起来。自然468 (7326): 927–932.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
19Qin S,Jin L,Zhang J,Liu L,Ji P,et al.(2011)通过溴代多巴胺-PHD指蛋白2的第一个PHD指识别未修饰的组蛋白H3,为组蛋白乙酰转移酶-单核细胞白血病锌指蛋白(MOZ)和MOZ相关因子(MORF)的调控提供了见解。生物化学杂志286 (42): 36944–36955.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
20Org T,Chignola F,Hetenyi C,Gaetani M,Rebane A,et al.(2008)自身免疫调节物PHD指与非甲基化组蛋白H3K4结合以激活基因表达。EMBO代表9 (4): 370–376.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
21.Koh AS、Kuo AJ、Park SY、Cheung P、Abramson J等(2008)Aire利用组蛋白结合模块调节免疫耐受,将染色质调节与器官特异性自身免疫联系起来。美国国家科学院程序105 (41): 15878–15883.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
22Peterson P,Org T,Rebane A(2008)AIRE的转录调控:中枢耐受的分子机制。Nat Rev免疫学8 (12): 948–957.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
23Koh AS、Kingston RE、Benoist C、Mathis D(2010),aire与组蛋白-3的低甲基化赖氨酸-4结合的全球相关性。美国国家科学院程序107 (29): 13016–13021.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
24.Heino M、Peterson P、Kudoh J、Shimizu N、Antonarakis SE等(2001),自身免疫调节器(AIRE)基因的APECED突变。Hum突变体18 (3): 205–211.[内政部] [公共医学] [谷歌学者]
-
25芬兰-德国APECED联合会(1997)一种自身免疫性疾病,APECED,由一种具有两个PHD型锌指结构域的新基因突变引起。芬兰-德国APECED财团。自身免疫性多内分泌病-念珠菌-表皮营养不良。自然基因17 (4): 399–403.[内政部] [公共医学] [谷歌学者]
-
26Akirav EM,Ruddle NH,Herold KC(2011)AIRE在人类自身免疫性疾病中的作用。Nat Rev内分泌7 (1): 25–33.[内政部] [公共医学] [谷歌学者]
-
27Gilson MK,Zhou HX(2007)《蛋白质-结合亲和力的计算》。生物物理生物分子结构年鉴36: 21–42.[内政部] [公共医学] [谷歌学者]
-
28霍S,Massova I,Kollman PA(2002)1∶1人类生长激素受体复合物的计算丙氨酸扫描。计算机化学杂志23 (1): 15–27.[内政部] [公共医学] [谷歌学者]
-
29Moreira IS,Fernandes PA,Ramos MJ(2008)《蛋白质识别:MDM2-P53复合物的计算突变研究》。Theor化学账户120 (4–6): 533–542.[谷歌学者]
-
30Bradshaw RT,Patel BH,Tate EW,Leatherbarrow RJ,Gould IR(2011)比较用于探测原型蛋白质-蛋白质相互作用的实验和计算丙氨酸扫描技术。蛋白质工程设计选择24 (1–2): 197–207.[内政部] [公共医学] [谷歌学者]
-
31Ivetac A,McCammon JA(2009)通过多拷贝分子动力学模拟阐明HIV-1非核苷逆转录酶抑制剂的抑制机制。分子生物学杂志388 (3): 644–658.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
32Amadei A,Linssen ABM,Berendsen HJC(1993)蛋白质的基本动力学。蛋白质17: 412–425.[内政部] [公共医学] [谷歌学者]
-
33Hayward S,de Groot L(2008)蛋白质的分子模型。正常模式和基本动力学。分子生物学方法443: 89–106.[内政部] [公共医学] [谷歌学者]
-
34Massova I,Kollman PA(1999),计算丙氨酸扫描以探测蛋白质相互作用:评估结合自由能的新方法。JACS公司121 (36): 8133–8143.[谷歌学者]
-
35Brown SP,Muchmore SW(2006)《蛋白质-甘氨酸结合亲和力的高通量计算:MM-PBSA协议对企业网格计算的修改和适应》。J Chem Inf模型46 (3): 999–1005.[内政部] [公共医学] [谷歌学者]
-
36Brown SP,Muchmore SW(2009),高通量分子力学与泊松-玻尔兹曼表面积的大规模应用,用于蛋白质-甘氨酸复合物的常规物理评分。化学52 (10): 3159–3165.[内政部] [公共医学] [谷歌学者]
-
37Yan C,Kaoud T,Lee S,Dalby KN,Ren P(2011)了解JNK1和支架蛋白JIP1之间对接相互作用的特异性。物理化学杂志B115 (6): 1491–1502.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
38Muzzioli E,Del Rio A,Rastelli G(2011)用分子动力学和mm-pbsa结合自由能计算评估蛋白激酶的选择性。化学生物药物设计78 (2): 252–259.[内政部] [公共医学] [谷歌学者]
-
39Lafont V,Schaefer M,Stote RH,Altschuh D,Dejaegere A(2007)抗原-抗体复合物中的蛋白质识别和相互作用热点:自由能分解识别“高效氨基酸”。蛋白质67 (2): 418–434.[内政部] [公共医学] [谷歌学者]
-
40Li Y,Li H(2012)《推动的许多关键:多样化植物同源域手指的“读者群”》。生物学报(上海)44 (1): 28–39.[内政部] [公共医学] [谷歌学者]
-
41Ozboyaci M,Gursoy A,Erman B,Keskin O(2011)通过JMJD2A的tudor结构域对H3/H4组蛋白尾部的分子识别:一项比较分子动力学模拟研究。公共科学图书馆一号6(3):e14765。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
42Arrowsmith CH、Bountra C、Fish PV、Lee K、Schapira M(2012)表观遗传蛋白家族:药物发现的新前沿。Nat Rev药物发现[内政部] [公共医学] [谷歌学者]
-
43Hess B、Kutzner C、van der Spoel D、Lindahl E(2008)《GROMACS 4:高效、负载平衡和可扩展分子模拟的算法》。JCTC公司4: 435–447.[内政部] [公共医学] [谷歌学者]
-
44Jorgensen W、Chandrasekhar J、Madura J、Impey R、Klein M(1983)模拟液态水的简单势函数比较。化学物理杂志79: 926–935.[谷歌学者]
-
45Amadei A,Ceruso MA,Di Nola A(1999)关于通过蛋白质分子动力学模拟的基本动力学分析获得的构象坐标基集的收敛性。蛋白质36 (4): 419–424.[公共医学] [谷歌学者]
-
46Baker NA,Sept D,Joseph S,Holst MJ,McCammon JA(2001)《纳米系统的静电:微管和核糖体的应用》。美国国家科学院程序98 (18): 10037–10041.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
47Brown SP,Muchmore SW(2009)具有泊松-玻尔兹曼表面积的高通量分子力学在蛋白质-配体复合物的常规物理评分中的大规模应用。化学52 (10): 3159–3165.[内政部] [公共医学] [谷歌学者]
-
48Gohlke H,Case DA(2004)《聚合自由能估算:MM-PB(GB)SA对蛋白质复合物ras-raf的研究》。计算机化学杂志25 (2): 238–250.[内政部] [公共医学] [谷歌学者]
-
49Kar P,Lipowsky R,Knecht V(2011)极性溶剂化对抗体及其变体与类固醇的交叉反应的重要性。物理化学杂志B115 (23): 7661–7669.[内政部] [公共医学] [谷歌学者]
-
50Cheatham TE 3rd,Srinivasan J,Case DA,Kollman PA(1998),polyG-polyC和polyA-polyT DNA双工体在溶液中稳定性的分子动力学和连续溶剂研究。生物分子结构动力学杂志16 (2): 265–280.[内政部] [公共医学] [谷歌学者]
-
51Majumdar R、Railkar R、Dighe RR(2011)使用重组抗体预测hCG-LH受体相互作用中涉及的构象域的对接和自由能模拟。蛋白质79 (11): 3108–3122.[内政部] [公共医学] [谷歌学者]
-
52Kumar R、Shinde RN、Ajay D、Sobhia ME(2010)《探索PTP1B抑制剂的相互作用要求:比较分子动力学研究》。J Chem Inf模型50 (6): 1147–1158.[内政部] [公共医学] [谷歌学者]
-
53Humpries W,Dalke A,Schulten K(1996)VMD:视觉分子动力学。J Mol图形14: 33–38.[内政部] [公共医学] [谷歌学者]
-
54Kabsch W,Sander C(1983)《蛋白质二级结构词典:氢键和几何特征的模式识别》。生物聚合物22 (12): 2577–2637.[内政部] [公共医学] [谷歌学者]
-
55Roberts E、Eargle J、Wright D、Luthey-Schulten Z(2006)MultiSeq:进化分析的统一序列和结构数据。BMC生物信息学7: 382.[内政部] [PMC免费文章] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。
补充材料
图S1
AIRE-PHD1/H3K4me0复合物的表面图。AIRE-PHD1(白色卡通和表面)和H3K4me0(橙色卡通)的综合体。与H3A1、H3R2、H3K4、H3R8和H3K9相互作用的AIRE-PHD1残基分别显示为绿色、品红色、青色、粉红色和紫色。虚线表示复合物的极性触点和Zn的选择2+离子用灰色球体表示。
(畅通节能法)
图S2
自由(青色)和束缚(蓝色)AIRE-PHD1的五个复制品的Cα原子从其时间平均位置的RMSF。
(TIFF)
图S3
AIRE-PHD1(残基295-344)和H3K4me0(残基1-5,黑线)的基于残基(Cα原子)的关联图。箭头表示AIRE-PHD1β1链与组蛋白尾部形成的额外β链之间的相关性。
(畅通节能法)
图S4
与中方框1、2和3所述相关性对应的相互作用
图2
.左侧显示了沿自由(青色)和束缚(蓝色)AIRE-PHD1动力学的特定距离分布,右侧显示了自由(青蓝色)和绑定(蓝色)空气-PHD1的两个代表性结构,灰色球体和黄色卡通表示Zn2+离子和组蛋白尾。(A) Glu296和Pro315主链原子之间的相互作用,(B)Glu298和Arg303侧链之间的盐桥,(C)Ser324和Ser338之间的氢键。
(畅通节能法)
图S5(A) Cα原子相对于不同系统模拟所得平均结构的RMSD值的累积平均值。(B) 不同系统五次模拟所得MM/PBSA值的累计平均值。
(畅通节能法)
图S6
CαRMSD来自(A)自由和(B)束缚启动AIRE-PHD1结构,作为50 ns轨迹中时间的函数。
(畅通节能法)
图S7
GMXAPBS工作流程示意图。
(畅通节能法)
表S1
MD模拟期间PHD手指和H3K4me0之间的原子间接触以及相关的相互作用能量。对每条轨迹的最后8纳秒进行分析。在该分析中,“接触”定义了在超过30%的总模拟帧中发生的任何一对原子之间的相互作用距离(<3º)。等效残留物,如图1所示的结构线形所定义图4C,与相应的能量贡献(kJ/mol)在同一条线上报告。
(文件)
表S2
自由和束缚AIRE-PHD1动力学的前六个特征向量捕获的总方差的方差比例和累积比例。
(文件)
表S3
用于MM/PBSA计算的PHD-H3K4me0络合物概要。
(文件)
表S4
自由和束缚AIRE-PHD1轨迹(#)的三个不同时间窗口(2-5、2-8和2-10 ns)获得的特征向量之间的RMSIP值。
(文件)
表S5
自由和束缚AIRE-PHD1轨迹(#)的三个不同时间窗口(2-10、2-30和2-50 ns)获得的特征向量之间的RMSIP值。
(文件)