结果和讨论
LEs和两小体在trpml公司1
果蝇属TRPML定位于培养细胞中的晚期终末小体/溶酶体[1]. 鉴定TRPML的亚细胞定位体内,我们表示UAS-trpml::myc使用镀锌4/无人机系统[2]. TRPML::MYC修饰LysoTracker阳性囊泡的外围(图1A–C),并与LE/溶酶体标记物YFP::Rab7共定位(图S1A–C)和LAMP::GFP(图1D–F). 这些数据表明,TRPML::MYC是一种LE/溶酶体膜蛋白,哺乳动物TRPML1也是如此[三].
图1。晚期内体和两性体的积累增加trpml公司1突变体。
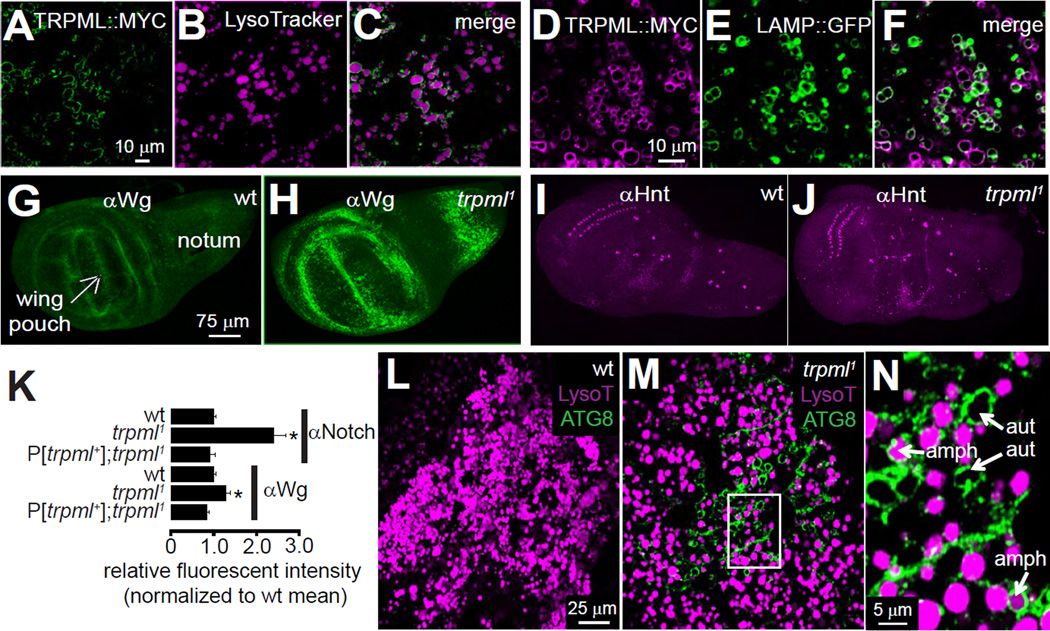
(A–C)TRPML::MYC(A,绿色)修饰LysoTracker阳性小泡(B,品红色)的外围体内.共聚焦图像3第个从表达UAS-trpml::myc在脂肪体特异性的控制下lsp2-GAL4号机组。使用抗MYC检测到TRPML::MYC定位。(C) (A–B)的合并图像。(A)中显示的比例尺适用于(A–C)。
(D–F)TRPML::MYC(D,品红色)与LAMP::GFP阳性囊泡共定位(E,绿色)体内.共聚焦图像3第个从表达UAS-trpml::myc在脂肪体特异性的控制下cg-GAL4公司。使用抗MYC检测到TRPML::MYC定位。(F) (D–E)的合并图像。(D)中显示的比例尺适用于(D–F)。
(G–J)野生型(wt)(G和I)和trpml公司1(H和J)3第个龄幼虫(96小时龄)翼盘染色:(G和H)抗无翼(αWg)、(I和J)抗后视(αHnt)和(N和O)抗缺口(Notch)。(G)中所示的比例尺适用于(G–J)。
(K) 无翼和缺口荧光强度增加的量化trpml公司1和P[trpml公司1];trpml公司1翼盘归一化为wt.值代表平均值±SEM;“*”表示p<0.05,学生t检验(n≥3)。
(L–M)从正常喂养的野生型(wt)(L)和trpml公司1(M) 徘徊3第个表达龄幼虫UAS-GFP::atg8在胖子司机的控制下cg-GAL4公司组织用抗GFP(ATG8,绿色)和LysoTracker(LysoT,品红色)染色。L中显示的比例尺也适用于M。
(N) (M)中方框区域的放大图像,显示自噬体(aut,仅抗GFP标记的小泡)和两性体(amph,抗GFP和LysoTracker标记的小囊)。
这个trpml公司1苍蝇无法完成自噬体的溶酶体降解[1]. 确定溶酶体降解途径中受影响的步骤trpml公司1,我们评估了果蝇属Wnt同源,无翼(Wg)[4]. Wg与受体结合后,内化到内体中,并在溶酶体中降解[5]. 我们发现Wg在翼囊和齿槽中的积累增加trpml公司1翼径(图1G-H和1K),该表型由trpml公司+基因组转基因(P[trpml公司+];trpml公司1) (图1K).
Wg可能在早期内体或LEs/多泡体(MVB)中积累trpml公司1光盘。为了区分这些可能性,我们依赖于Wg在质膜和早期内体传递信号的观察结果。只有在MVB形成后,信号才终止。因此,Wg信号在trpml公司1这表明Wg在早期内体中积累,而Wg信号的不变与Wg在MVB中的积累一致。因此,我们评估了Wg靶基因Hindsight(Hnt)在翼状突起中的激活情况,如所述[4]. 细胞核Hnt的表达在野生型和trpml公司1(图1I–J)表明Wg信号在trpml公司1.
我们还使用Notch内吞域特异性抗体评估了Notch的积累,发现Notch水平在trpml公司1翼片(图S1D–E和1K). 缺口水平似乎高于Wgtrpml公司1因为当Wg积聚在翼囊和凹槽中时,凹槽在整个椎间盘上升高。支持以下结论:trpml公司1是LEs,Notch阳性小泡与LysoTracker共同染色(图S1F–H).
自噬是一种降解细胞大分子所需的途径,这些大分子太大,无法通过蛋白质体桶[6,7]. 在自噬过程中,被称为自噬体的双膜结合囊泡分离了注定要降解的胞质物质。随后,自噬体与LEs/MVB融合形成两性体[8,9]. Amphisome随后与溶酶体结合,形成自溶体。由于溶酶体携带降解酶,在自溶酶体形成后,两性体的内容物被分解[8,9].
我们之前报道过trpml公司1成人表现出自噬流量减少的特征[1]. 为了提供自噬体和两性体积聚的证据,我们对野生型和trpml公司1使用LysoTracker和GFP的脂肪体::ATG8。自噬体仅用GFP::ATG8标记,而两性体同时用GFP::ATG5和LysoTracker染色。尽管野生型几乎没有GFP::ATG8染色(图1L),有很多trpml公司1仅用GFP::ATG8标记的囊泡或同时使用GFP::ATG8和LysoTracker标记的囊囊泡(图1M–N). 这些数据表明trpml公司导致自噬体和两性体升高。在缺乏TRPML1的人类细胞中以及在秀丽线虫一个突变破坏了蠕虫TRPML1同源物[10——12]. 这些trpml公司1表型与缺乏工厂1(囊泡磷脂酰肌醇3-磷酸5-激酶)[4]. 这些表型相似性与哺乳动物TRPML1被Fab1/PIK-FYVE-激酶、PI(3,5)P(2)的产物激活的发现一致[13].
两性体与溶酶体融合的缺陷
我们还对幼虫脂肪体进行了电镜检查,以根据先前描述的形态学标准确认积聚囊泡的性质[14]. 野生型脂肪体细胞含有大的电子密度溶酶体样结构(图2A和C). 许多溶酶体处于融合过程中,呈“图8”样结构,没有内部分离(图2A和蓝色线条图2C). 相反,trpml公司1脂肪体细胞包含的溶酶体越来越少,越来越小(图2B). 这种表型类似于深橙色(多尔)突变脂肪体,其特征是LEs与溶酶体的融合减少[14]. 此外,trpml公司1突变细胞积累了大的单膜结合囊泡,其中包含较小的内部囊泡,表明它们是MVB/两性体(图2B和D–E). 许多囊泡接触到溶酶体,显然无法融合(图2D-E). 我们认为,这些“融合夹持”小泡是由MVB和/或两性体与溶酶体融合的缺陷引起的trpml公司1. Thetrpml公司1脂肪体也显示融合夹持溶酶体(图2F)表明囊泡的同型和异型融合均被破坏。
图2。囊泡融合缺陷的超微结构分析trpml公司1.
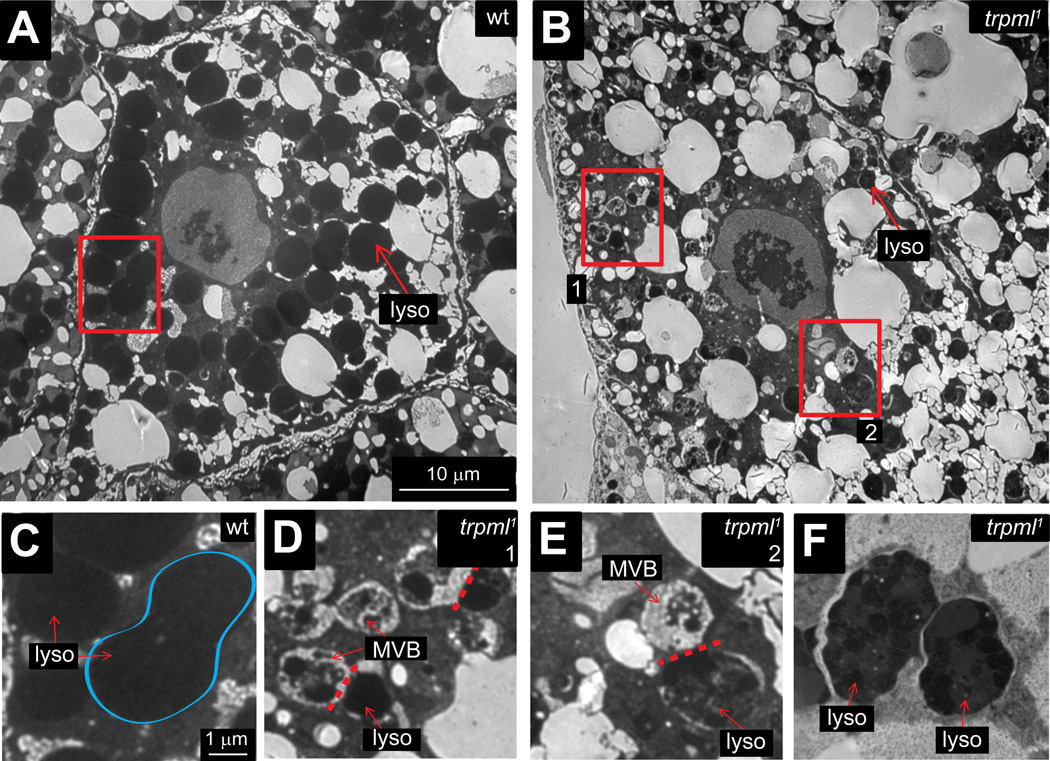
(A–B)正常喂养的野生型(wt)的代表性透射EM图像(A)和trpml公司1(B) 漫游3第个龄幼虫脂肪体。(A)中所示的比例尺也适用于(B)。
(C) 方框区域的放大图像,如(A)所示。蓝线表示融合过程中有两个溶酶体。(C)中显示的比例尺也适用于(D–F)。
(D) (B)中的方框1。
(E) (B)中的方框2。
(F) 放大图像显示两个融合夹持的溶酶体。
标记的结构包括溶酶体(lyso)和多泡体(MVB)。红色虚线表示“融合夹持”小泡之间的接触区域。
量化增加的囊泡积聚trpml公司1,我们对成人感光细胞(PC)进行了EM,与脂肪体细胞相比,后者更容易量化,因为前者的细胞边界更明显。我们发现,虽然野生型PC在每个切片中显示出极少的MVB(具有内部囊泡结构的单膜结合囊泡)和溶酶体(电子致密多层膜结构),trpml公司1PC显示MVB和溶酶体显著升高(图S2A-B和S2G). 突变细胞也显示自噬体的积累增加(含有胞质物质的双层膜囊泡)(图S2G). 成人PC中较高水平的自噬体与幼虫脂肪体中的发现一致。MVB和溶酶体融合后形成的自体溶酶体的相对数量(包含内部囊泡结构的单膜结合囊泡和多层电子致密溶酶体)在野生型和trpml公司1细胞,尽管两倍体和溶酶体增加(图S2G). 此外,MVBs/自体溶酶体的比率在trpml公司1与野生型相比(分别为11.7和1.2;图S2G). 这个trpml公司1–PC中还含有许多接触溶酶体的MVB(2.2±0.8融合夹持囊泡/小泡trpml公司1突变体)(图S2C–G). 野生型细胞中这些“融合夹持”小泡的数量显著低于野生型细胞(0.1±0.1融合夹持小泡/小泡,p=0.04,Student t检验)(图S2G). 综上所述,我们的数据表明MVB与溶酶体融合存在缺陷trpml公司1细胞。此外,由于突变体中自溶体的总数没有变化,我们认为它们在溶酶体降解方面表现出额外的缺陷。
LE-Ca升高2+在中trpml公司1突变体
哺乳动物TRPML1是负责腔钙释放的LE/溶酶体通道2+[13,15,16]. 因此,TRPML1的缺失可能导致LE-Ca升高2+水平。调查是否消除果蝇trpml导致LE Ca升高2+水平,我们测量了LE管腔Ca的释放池2+Fura-2脂肪体。用于释放LE Ca2+我们用巴非霉素A1(一种H型液泡的细胞发酵抑制剂)处理脂肪体+-ATP酶。巴非霉素A1治疗导致LE H损失+LE Ca的梯度和最终耗尽2+,增加细胞溶质游离钙2+浓度[17]. 与以下概念一致:trpml公司1突变体使LE-Ca升高2+bafilomycin A1处理导致胞浆游离钙显著升高2+在里面trpml公司1与野生型相比(重量为0.03±0.006和0.15±0.05trpml公司1分别)(图S3A和图3A-B). 这些数据表明trpml公司1LEs含有较高水平的Ca2+由于钙减少2+释放。
图3。溶酶体钙升高2+,自噬诱导增加,TORC1信号减少trpml公司1突变体。
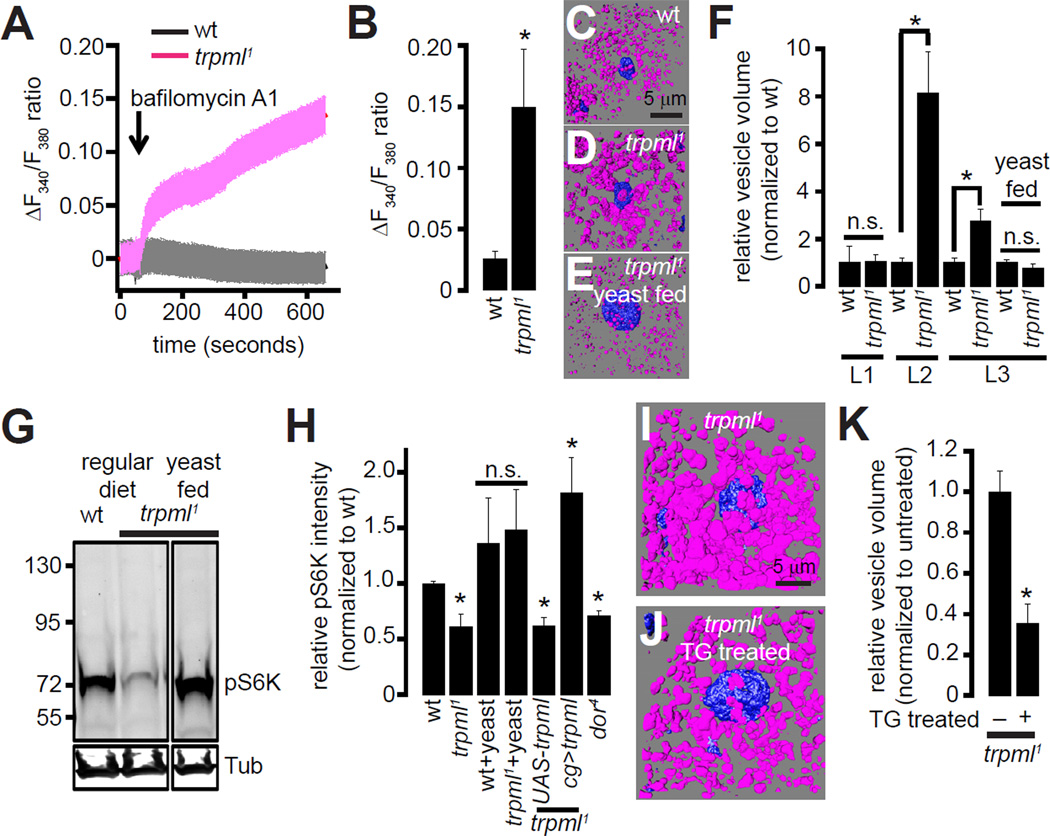
(A) 显示F整体变化的图表340/F类380重量比(黑色)和trpml公司1(洋红)纸巾。箭头表示添加巴非霉素A1的点。数值代表每个基因型的平均值±SEM,n~70个细胞(7个单独实验,每个实验约10个细胞)。
(B) 显示Fura-2 F变化的条形图340/F类380从t=0秒到t=600秒的比率。值表示平均值±SEM。“*”表示p<0.05,即Student t检验(n=7个实验/基因型)。
(C–E)400 mm的体积三维重建2显示LysoTracker(洋红色)和DAPI(蓝色)染色的区域(20 mm×20 mm)3第个指示基因型和摄食状态的龄幼虫脂肪体。(C)中所示的比例尺适用于(C–E)。
(F) 脂肪体中LysoTracker阳性囊泡的体积从1标准, 2第和3第个instar wt和trpml公司1幼虫(分别为L1-L3)。将每个幼虫阶段的值归一化为重量。数值代表平均值±SEM。“*”表示p<0.05,Student t检验(每个基因型的动物数量为5–7只)。
(G) 野生型(wt)和trpml公司1和酵母饲料trpml公司1三第个用磷酸化S6K(pS6K)和α-微管蛋白(Tub)抗体探测龄幼虫。
(H) 游荡3脂肪体提取物中相对pS6K强度的量化第个指示基因型和摄食状态的龄幼虫。将数值归一化为重量。数值代表平均值±SEM。“*”表示p<0.05,Student t检验(n=5-9个独立的Western blots,每次使用不同的样本)。术语cg公司指cg-GAL4公司,一种脂肪体和血细胞特异性驱动因素。
(I–J)400 mm的三维体积重建2显示LysoTracker(洋红色)和DAPI(蓝色)染色的区域(20 mm×20 mm)3第个龄期trpml公司1幼虫脂肪体。(J) 显示thapsigargin(TG)治疗后LysoTracker阳性小泡。(I)中所示的比例尺适用于(I–J)。
(K) 显示3个脂肪体中LysoTracker阳性小泡相对体积的条形图第个龄期trpml公司1未经处理或经TG处理的幼虫。值按未经处理的样本标准化。数值代表平均值±SEM。“*”表示p<0.05,Student t检验(n=9只每种基因型的动物)。
TORC1活性降低导致自噬诱导增加trpml公司1
我们比较了野生型和野生型LysoTracker阳性囊泡的大小trpml公司1幼虫脂肪体评估自噬诱导[18]. 与自噬诱导增加一致trpml公司1,突变脂肪体中LysoTracker阳性小泡的体积显著增加(图3C–D和F). 这种变化变得明显,在2岁以后的脂肪体中最为明显第龄幼虫(8.14±1.7倍trpml公司1) (图3F). 野生型和trpml公司1不那么明显,但在3第个龄幼虫(增加2.77±0.5倍trpml公司1) (图3F). 中的较小标高trpml公司1三第个龄幼虫可能反映了在这个发育阶段野生型组织中蜕皮激素依赖的自噬激活[14,19].
我们假设,由于两个因素,在未完成自噬的情况下诱导自噬应该会抑制TORC1活性。首先,自噬通量的下降会降低蛋白质自噬降解产生的氨基酸的净可用性[20]. 氨基酸水平降低会降低T型目标R(右)阿帕霉素C类复合体1(TORC1)[21——23]. 事实上,Atg7敲除导致的自噬流量减少导致TORC1活性降低,这是由TORC1底物S6-激酶(pS6K)的磷酸化决定的[21,24]. 我们发现在野生型脂肪体中使用RNAi敲除Atg5后,TORC1活性也有类似的下降(图S3B–C). 其次,自噬增加将直接抑制TORC1功能,因为自噬和TORC1活性是相互拮抗的[24]. TORC1降低将进一步诱导自噬,导致产生更大的LysoTracker阳性囊泡。
有几条证据支持上述建议。首先,喂食trpml公司1三第个龄幼虫富含蛋白质的酵母糊抑制了LysoTracker阳性囊泡体积的增加(图3E–F). 其次,S6激酶的磷酸化在trpml公司1脂肪体(图3G–H). pS6K的减少trpml公司1通过驾驶野生型被逆转trpml公司+在脂肪体中使用cg公司-镀锌4[25] (图3H). 此外,酵母喂养抑制了pS6K水平的降低trpml公司1(图3G–H). 为了研究其他LEs与溶酶体融合减少的突变体中TORC1活性是否降低,我们评估了多尔突变体[14,26]. 摘录自多尔4突变幼虫的pS6K降低(图3H). 因此,LEs与溶酶体融合的阻断导致细胞氨基酸水平降低和TORC1活性降低。
第三,通过过度表达Rheb和组成活性Rag(RagQ61L问题) [27,28]LysoTracker阳性囊泡体积减少(图S3D). 囊泡体积trpml公司1当我们表达显性负Rag(RagT16N型) [27]表明Rag活性在trpml公司1发现提高TORC1活性足以抑制溶酶体的储存,这表明trpml公司1反映了TORC1活动的减少。因此,尽管囊泡融合缺陷持续存在,但提高TORC1活性足以防止囊泡积聚。
第四,化蛹的半最大时间在trpml公司1(图S3E). 由于TORC1活性降低导致发育延迟[29],这些数据也与TORC1活性在trpml公司1.喂食幼虫富含蛋白质的酵母糊将pS6K水平恢复到野生型(图3G–H)挽救了发展时机的缺陷(图S3E).
LE/溶酶体钙2+是这些囊泡同型和异型融合所必需的[30]. 因此,LysoTracker染色增加trpml公司1可能是由于钙减少所致2+小泡的释放导致LEs/两性体与溶酶体的融合受损,最终导致其内容物降解减少。与建议一致,水泡体积的增加源于钙的减少2+释放并用他司加金来治疗脂肪体,这会阻塞SERCA泵并导致Ca2+从内质网储存中释放,导致LysoTracker阳性囊泡的体积显著减少trpml公司1(图3I–K). 因此,尽管缺乏LE-Ca2+中的释放机构trpml公司1,胞浆钙升高2+不同Ca的水平2+储备足以部分抑制LysoTracker阳性囊泡体积的增加。虽然我们的数据与trpml公司1,我们不能排除囊泡运输中也可能存在缺陷,从而减少易熔囊泡之间的接触。
抑制trpml公司高蛋白饮食的半致死性
蛹期果蝇属不进食,依赖自噬获取形态发生和生存所必需的氨基酸。损失trpml公司在蛹期导致半致死,因为<10%的成年人从蛹病例中羽化(图4A和S4C系列) [1]. 为了测试这种活力下降是否是由于氨基酸供应不足造成的,我们给突变幼虫喂食高蛋白食物(添加20%w/v酵母的食物)。我们发现这种饮食显著抑制了致命性(图4A和第4c页). 然而,另一种导致蛹致死的突变(P元件插入vamp-7型,CG1599号EY09354(眼睛09354);图S4A–B)不受酵母补充物的抑制(图S4C).
图4。通过喂食富含蛋白质的食物来激活TORC1,可以抑制与失去trpml公司1.
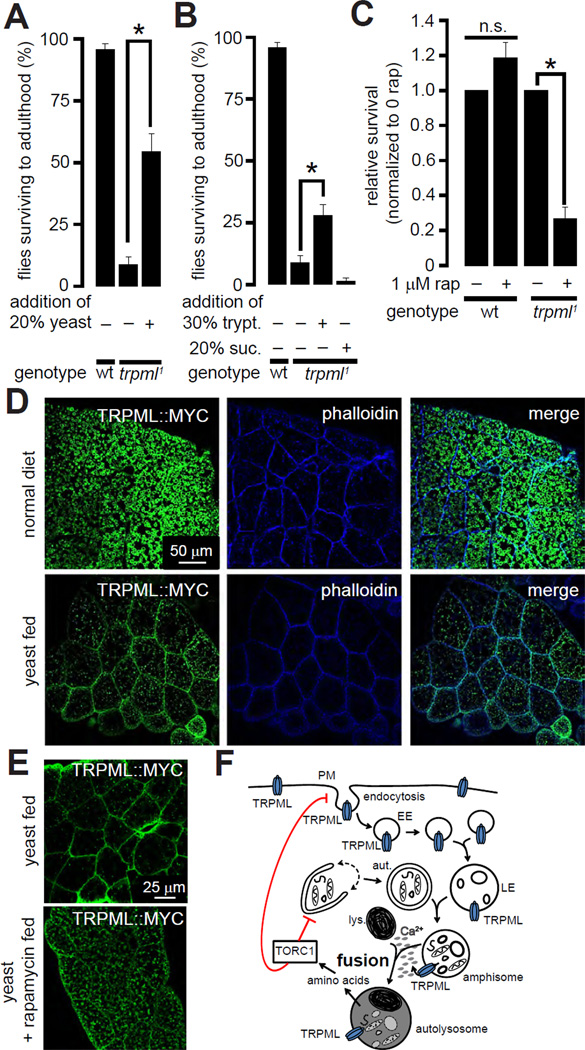
(A) 条形图显示了在标准食物或含有20%酵母的食物中饲养的指定基因型苍蝇的存活率。数值代表平均值±SEM。“*”表示p<0.05,学生t检验(n≥3个独立飞瓶)。
(B) 显示用标准食物或含有指定额外饲料的食物(trypt)饲养的指定基因型苍蝇存活率的条形图。(胰蛋白胨)或类似物。(蔗糖)。数值代表平均值±SEM。“*”表示p<0.05,ANOVA,然后是带有Bonferroni事后校正的成对Student t检验(n≥3个独立飞行瓶)。
(C) wt和trpml公司1在存在或不存在1µM雷帕霉素(rap)的情况下,在食物中饲养时,动物经过蛹期。这些值被归一化为不使用雷帕霉素饲养的苍蝇的存活率。数值代表平均值±SEM。“*”表示p<0.05,学生t检验;n.s.显示无显著性(n≥3个独立的飞行小瓶)。
(D) 共聚焦图像显示3例脂肪体中的TRPML::MYC(绿色)和卵磷脂(蓝色)染色第个表达龄幼虫UAS-trpml::myc在cg-GAL4公司显示了从酵母喂养的幼虫身上切下的组织。左上方面板中显示的比例尺适用于所有面板。
(E) 共聚焦图像显示3例脂肪体中的TRPML::MYC染色第个表达龄幼虫UAS-trpml::myc在…的控制下cg-GAL4公司显示了从酵母饲料和酵母+雷帕霉素饲料的幼虫上切下的组织。左侧面板中显示的比例尺适用于两个面板。
(F) TRPML调节内体运输和TORC1活性的功能模型。PM,质膜;EE,早期内体;LE,迟发型;aut,自噬体;溶酶体。
抑制trpml公司酵母糊的半致死性可能是由于该补充剂中的蛋白质或碳水化合物所致。因此,我们测试了补充胰蛋白胨或蔗糖是否会降低致死率。我们发现,虽然补充胰蛋白胨降低了半致死性,但补充蔗糖没有降低半致死性(图4B和S4D系列). 缺乏蔗糖抑制表明,该表型不是热量缺乏的结果,而是反映了通过以下方式增加饮食氨基酸的需求trpml公司1幼虫。
接下来,我们考虑了高蛋白饮食对半致死性的抑制是否是由于TORC1活性增加所致。我们用酵母糊trpml公司1在TORC1-抑制剂雷帕霉素存在下的幼虫。我们发现雷帕霉素阻止了酵母糊对蛹半致死性的抑制(图S4E). 此外,雷帕霉素在trpml公司1幼虫是用普通食物喂养的(图4C). 然而,为多尔突变体雷帕霉素并没有降低其生存能力(数据未显示)。这些数据表明,并非所有LEs与溶酶体融合不足的突变体都表现出对雷帕霉素的敏感性增加。
TORC1同时增加蛋白质翻译并减少自噬。TORC1增加蛋白质翻译的途径之一是磷酸化和抑制4E-BP1的翻译抑制因子Thor-fly同源物[31,32]. 因此,如果TORC1激活的影响trpml公司1依赖于蛋白质翻译,雷帕霉素不应逆转酵母在雷神2;trpml公司1双突变体动物。然而,酵母糊仍然抑制了雷神2;trpml公司1双突变体(无雷神2;trpml公司1成年人在没有添加酵母的饮食中闭关),雷帕霉素的作用保持不变(图S4E). 这些结果表明,高水平氨基酸激活TORC1可能抑制了蛹的半致死性trpml公司1通过减少自噬而不是增加蛋白质翻译。
TORC1调节TRPML的亚细胞定位
为了研究TORC1活性是否相互影响TRPML,我们检测了在我们操纵TORC1活性的条件下TRPML::MYC的空间分布。在正常饮食中,TRPML::MYC完全是细胞内的(图4D). 然而,在高蛋白饮食中,TRPML::MYC与皮质F-肌动蛋白标记物卵磷脂共定位(图4D)表明TRPML位于质膜(PM)。在维持高蛋白饮食和雷帕霉素的幼虫中,我们仅在细胞内小泡中检测到TRPML::MYC(图4E). 这些数据表明,TRPML::MYC的PM定位取决于TORC1的活性。进一步支持这一结论,TRPML::MYC主要定位于幼虫唾液腺中的PM(图S4F)其特征是自噬水平低(因此TORC1活性高),直到蛹期开始,此时自噬是变态过程中蛹唾液腺降解所必需的[33].
TORC1活性对TRPML亚细胞定位的影响不太可能反映大量内吞作用的改变,因为TORC1增强而不是抑制大量内吞[34,35]. 相反,我们建议通过阻断TRPML进入内体并降低LEs中TRPML的水平,TORC1对自噬的完成起反馈调节作用。因此,除了抑制自噬的启动外[24],TORC1还通过调节TRPML的亚细胞位置来抑制自噬的完成。
TRPML功能模型和结论
TRPML是Ca2+通道,从PM内吞并最终进入LEs(图4F). LEs与自噬体融合,产生两性体。两性体中存在的TRPML释放管腔钙2+促进钙2+两性体与溶酶体的依赖性融合。自溶体中蛋白质降解产生的氨基酸促进TORC1活化。除了抑制自噬的启动外,激活的TORC1还减少TRPML的内吞作用。
在缺乏TRPML的情况下,两性体和溶酶体的融合受到损害。这导致氨基酸自噬流量减少,导致TORC1减少和自噬上调。补充trpml公司1富含蛋白质酵母的饮食可以逆转TORC1活性降低的影响。这些发现增加了MLIV患者TORC1活性降低的可能性。如果是这样,那么很有意思的推测是,补充氨基酸可能会降低MLIV相关临床表现的严重程度。

