背景:精氨酸琥珀酸合成酶(AS)对内皮细胞一氧化氮的生成至关重要,但对其调节机制知之甚少。
结果:AS Ser-328磷酸化随着钙刺激而增加,而随着PKCα的干扰而降低。
结论:PKCα在钙依赖性刺激条件下磷酸化Ser-328处的AS,以支持一氧化氮的生成。
意义:了解AS是如何调节的,对于理解一氧化氮稳态至关重要。
关键词:内皮细胞、一氧化氮、一氧化氮合酶、磷酸化、蛋白激酶C(PKC)、精氨酸琥珀酸合酶
摘要
内皮一氧化氮合酶(eNOS)利用我-精氨酸作为其主要底物,将其转化为我-瓜氨酸和一氧化氮(NO)。我-瓜氨酸回收至我-精氨酸由精氨酸琥珀酸合成酶(AS)和精氨酸丁二酸裂解酶两种酶合成,为内皮细胞中eNOS和NO的生成提供底物精氨酸。这三种酶eNOS、AS和精氨琥珀酸裂解酶共同构成瓜氨酸-NO循环。虽然AS可以催化NO生成的速率限制步骤,但对于AS在内皮细胞中转录水平以外的调节却知之甚少。在本研究中,我们发现,当牛主动脉内皮细胞被钙离子载体或他司加金刺激产生NO时,AS Ser-328磷酸化与eNOS Ser-1179磷酸化协同调节在体外激酶分析、激酶抑制研究以及蛋白激酶Cα(PKCα)敲除实验表明,as Ser-328的钙依赖性磷酸化是由PKCα介导的。总的来说,这些发现表明,在促进NO生成的条件下,根据eNOS的钙依赖性调节,调节Ser-328处AS的磷酸化,并与AS在血管内皮细胞瓜氨酸-NO循环中的速率限制作用相一致。
介绍
一氧化氮(NO)的产生在血管内皮细胞中受到严格调控。这种控制的损害与损害内皮功能的风险因素有关。精氨酸导向eNOS2为了维持NO的生成,瓜氨酸再循环为精氨酸,在内皮细胞中形成一个独特的精氨酸池(1–4). 这种从大量胞液精氨酸中分离出来的细胞内精氨酸池是通过精氨酸循环酶精氨琥珀酸合成酶(AS)和精氨琥酯裂解酶的作用生成的,AS的催化作用是内皮细胞NO生成的速率限制(1).
由于内皮细胞代表了一个独特的环境,在整合对生理线索的反应的信号级联的持续影响下,有必要对AS进行急性或立即调节,以适应eNOS已知的调节反应。例如,研究表明,VEGF和胰岛素信号能够协调调节eNOS的两个磷酸化位点,增加Ser-1179的磷酸化,降低Thr-497的磷酸化水平,从而影响eNOS活性的整体增加(6). 这种由VEGF或胰岛素信号引起的eNOS磷酸化的变化主要由钙非依赖性激酶介导,如Akt(7).
尽管最近的研究表明AS是一种磷酸蛋白(8,9),没有生理相关的磷酸化位点的报道。2008年,一项自动化磷酸蛋白组分析显示,AS在HeLa细胞中的丝氨酸352处被磷酸化,但其生理意义尚未确定(9). 同年晚些时候,科尔宾等。(8)还证明AS是一种磷酸蛋白,VEGF刺激NO生成通过PKA途径影响AS的磷酸化,但未发现磷酸化位点。在本研究中,我们现在表明,在Ser-328处AS的磷酸化是对eNOS钙依赖性激活的反应,并且由PKC的α同型(PKCα)介导。
实验程序
细胞培养
如参考文献。10经过修改。简单地说,采集主动脉并用不含镁或钙的PBS清洗以清除血液,通过纵向解剖暴露管腔表面,然后用0.1%胶原酶孵育10分钟。内皮细胞分离是通过在表面滚动无菌拭子实现的,然后通过在5 ml富含Dulbecco改良Eagle's培养基(DMEM,1 g/L葡萄糖,Invitrogen)中旋转拭子释放细胞,该培养基含有10%FBS(HyClone)、100 IU/ml青霉素和链霉素以及250 ng/ml两性霉素B(Cellgro)在15-ml锥形管中。将细胞制成颗粒,重新悬浮在6 ml DMEM+10%FBS中,然后在25 cm内进行电镀2处理过的细胞培养瓶。流式细胞术证实了培养中内皮细胞的分离。
在治疗之前,将汇合的BAEC(在第3代和第9代之间)在补充有0.1%BSA和1mL BSA的DMEM(1g/l葡萄糖,减去酚红,Invitrogen)中血清饥饿16小时米谷氨酰胺。治疗包括100 n孵育米胰岛素(西格玛),100 ng/ml VEGF(研发系统),0.5μ米钙离子载体A23187(Sigma),10μ米缓激肽(西格玛),42μ米罗特林,或2.5μ米用于PKC抑制或50μ米BAPTA-AM(钙生物化学)。孵化时间如图例所示。对照组仅用血清饥饿培养基治疗。
AS变异体的产生和瞬时转染
将AS cDNA克隆到pcDNA 3.1/V5-His,B表达载体(Invitrogen)中。然后,按照制造商的说明,通过QuikChange试剂盒(Stratagene)对AS构建物进行定点突变。简言之,Ser-328突变为丙氨酸以模拟非磷酸化状态,突变为天冬氨酸以模拟组成磷酸化状态。用于S328A突变的引物是有意义的:ACGGGTTTCTGGCACGCGCCCGAGTGAATTT和反义的:AAATTCACTCGGCGCTGCCAGAACCCGT。用于S328D突变的引物是有意义的:ACGGGTTTCTGGCACGACCGAGTGAATTT和反义的:AAATTCACTCGGGTCGTGCCAGAACCCGT。使用Lipofectamine 2000(Invitrogen)在无血清Opti-MEM I(Invit罗gen)中瞬时将实验质粒(6孔培养皿中每孔1μg DNA)转染到BAEC中。4 h后,用含10%血清的DMEM替换培养基,培养细胞6或24 h。用钙离子载体A23187和原钒酸钠刺激细胞4 h,然后通过2,3-二氨基萘(DAN)测定培养基中NO的生成。
质谱分析
用AS瞬时转染的BAEC孵育24小时,血清饥饿16小时,并用10μ米缓激肽或50 n米冈田酸作用30分钟。过表达的AS通过其融合的His纯化6使用镍-硝基三乙酸-海藻糖磁珠标记(Qiagen)。用SDS-PAGE分离蛋白质以鉴定AS带(51 kDa)。使用重复凝胶通过Western blot确认AS质粒的表达。将感兴趣的凝胶带切除并去除污渍。蛋白质二硫化物用三羧乙基膦还原,半胱氨酸用碘乙酰胺烷基化。采用凝胶胰蛋白酶消化法进行蛋白质水解。采用纳米流反相液相色谱法通过疏水性分离肽(LC Packings,Dionex,Sunnyvale,CA)。使用电喷雾线性离子阱质谱仪(LTQ,Thermo Scientific)完成在线检测。使用Mascot和SEQUEST数据库搜索算法将串联质谱分配给肽序列。通过手动检查数据来验证序列分配。
精氨酸琥珀酸合成酶磷酸丝氨酸Ser-328特异性抗体
21世纪生物化学公司使用磷酸肽AS序列322–335:RR-Ahx-YGFWH[pS]PECEFVR-amide(其中pS表示磷酸)生成了针对Ser(P)-328 AS的磷酸特异性抗体。通过进行磷酸肽竞争和免疫沉淀,然后进行Western印迹,确定磷酸-AS抗体的特异性。
Western Blot分析
在添加蛋白酶和磷酸酶抑制剂(Pierce)的放射免疫沉淀分析缓冲液中裂解BAEC,然后刮取收集。对全细胞裂解物进行澄清,并使用BCA蛋白测定法(Pierce)对总蛋白进行定量。等量(20μg)的蛋白质在4–15%Tris-HCl TGX预制凝胶(Bio-Rad)上溶解,并印在Immobilon-P聚偏氟乙烯膜上。如前所述进行蛋白质印迹(11). 在100%StartingBlock(Pierce)中封闭膜,并在20%StartingBlock1:2500抗AS(BD Transduction Laboratories)中与一级抗体孵育。使用的二级抗体是1:50000稀释度的过氧化物偶联山羊抗鼠或抗兔IgG(Jackson ImmunoResearch Laboratories)。使用West Pico试剂(Pierce)通过化学发光对斑点进行可视化,并将其暴露于薄膜中。使用Quantity-One软件(Bio-Read)对谱带强度进行量化。
一氧化氮测定
如前所述,使用荧光DAN(Sigma)分析法测量培养基中的亚硝酸盐,作为细胞NO的指示剂(12). 在96 well黑板中进行反应,在BMG FLUOstar Galaxy荧光光谱仪板读卡器上读取荧光,在360 nm激发,在405 nm检测发射。数据描述为每微克蛋白质中亚硝酸盐的pmol。
PKCα敲除试验
将90%融合的BAEC与靶向PKCα第五外显子(集成DNA技术)的dicer底物siRNA(DsiRNA)转染48小时,使用Lipofectamine 2000(Invitrogen),如前所述进行as构建。用于沉默基因表达的双链序列如下:5′-CGAGGAGCAAGCACAAGUUCAAGAT-3′和5′-AUCUGAACUGUGUGCUCGGG-3′。然后用钙离子载体(A23187,Sigma)处理转染的BAEC,并在蛋白印迹后进行裂解。如上所述,对所示蛋白质的相对量进行定量。
体外激酶检测
按照科尔宾的描述进行分析等。(8). 简单地说,纯化的重组AS是通过牛AS亚克隆产生的(NP_776317型)转化为pET-28(c)+载体(Novagen),并用大肠杆菌按照制造商的说明(Novagen),随后通过His·Bind树脂纯化蛋白质。SDS-PAGE证实纯化成功。用于测定每个激酶激酶活性的阳性对照含有适用的肽底物,而阴性对照含有除肽以外的所有成分。添加[33P] 每种反应混合物的ATP启动了在30°C下培养30分钟的分析。通过在磷纤维素P81板上进行反应斑点,然后进行三次清洗,可以终止分析,然后通过闪烁计数读取放射性。图像在体外通过添加样品缓冲液终止激酶反应,然后在10%聚丙烯酰胺凝胶上分馏。
结果
精氨酸琥珀酸合成酶Ser-328磷酸化位点的鉴定
为了在AS序列中建立具有生物学意义的磷酸化位点,我们通过应用质谱(LC-MS/MS)对经缓激肽或冈田酸(蛋白磷酸酶2A(PP2A)和PP1磷酸酶抑制剂)处理的培养BAEC纯化的过表达AS进行了蛋白质组学分析。质谱分析揭示了三个高度保守的丝氨酸/苏氨酸磷酸化位点,它们与背景离子信号不同。其中包括Thr-131/Ser-134、Ser-180/Ser-189和Ser-328(图1). Thr-131与Ser-134的接近性以及Ser-180与Ser-189的接近性使得这些单独的信号无法区分,因此被描述为Thr-131/Ser-134和Ser-180/Ser-189。
图1。
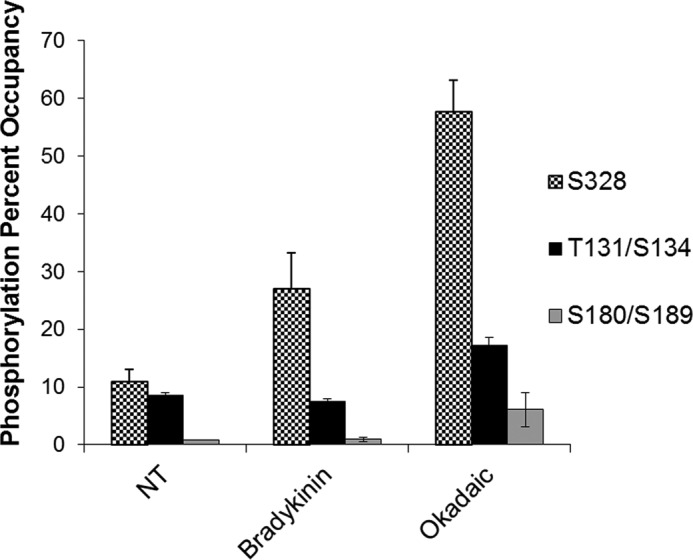
328系列(328件)是激酶可接近的暴露位点,并且显示出相对于治疗的磷酸化变化。用AS表达载体瞬时转染BAEC,然后用10μ米缓激肽或50 n米冈田酸。AS是他的标签纯化,然后进行SDS-PAGE。在串联质谱分析之前,切除与AS对应的带并消化胰蛋白酶。结果代表一个实验,用三份样本进行分析,S.E.表示为错误栏。NT公司,未处理。
在确定的位点中,Ser-328在用磷酸酶抑制剂(冈田酸)处理的细胞和未处理的细胞之间显示出最大的信号变化。此外,与未经处理的对照细胞相比,经缓激肽处理的Ser-328的信号发生了实质性变化(图1). 此外,生物信息学建模表明,激酶相互作用最容易到达的位点是Thr-131/Ser-134和Ser-328,而Ser-180/Ser-189似乎埋藏在酶的活性位点附近(数据未显示)。根据这些结果,Ser-328被选为磷酸化最强的候选基因体内因此,开发了一种针对Ser(P)-328的磷酸特异性抗体用于进一步研究(见“实验程序”)。
钙依赖性刺激BAEC降低Ser-328的磷酸化
为了确定Ser-328的磷酸化是否具有生理意义,通过改变NO生成的生理线索来检查Ser-328磷酸化相对于治疗的变化。特别是,选择VEGF和胰岛素刺激NO生成有两个原因。首先,有证据表明,血管内皮生长因子(VEGF)治疗后AS磷酸化发生改变,而VEGF治疗是通过PKA信号介导的(8). 第二,已知VEGF和胰岛素通过促进eNOS的Ser-1179磷酸化来刺激内皮细胞NO的生成(13,14).
如所示图2,与文献一致(13),我们发现胰岛素和VEGF促进Ser-1179处约50%的eNOS的磷酸化增加(Western印迹,图2,A类和C; 密度测定,图2,B类和D类)NO生成相应增加(图2E类). Thr-497处eNOS的磷酸化也随之降低约40%(Western blot,图2,A类和C; 密度测定,图2,B类和D类) (15–17). 令人惊讶的是,通过VEGF(85%)或胰岛素(40%)治疗,Ser-328的磷酸化显著降低(Western blot,图2,A类和C; 密度测定,图2,B类和D类).
图2。
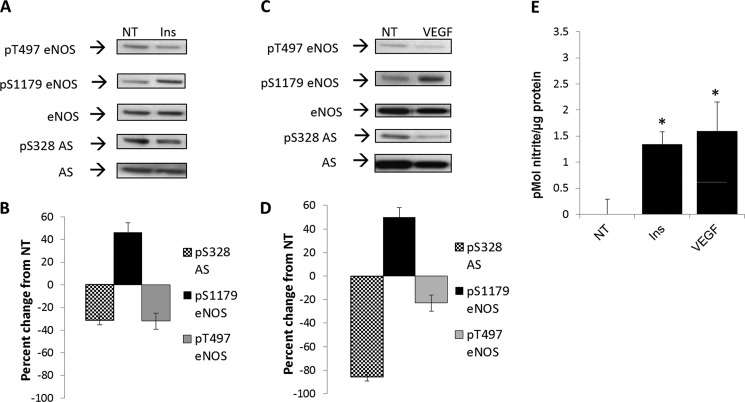
钙诱导的NO生成依赖性刺激降低精氨琥珀酸合成酶Ser-328的磷酸化(328件).
A类和C,BAEC接受胰岛素治疗(Ins公司)在100 n时米(A类)或100 ng/ml时的VEGF(C)20分钟。磷酸化(P(P))使用磷酸特异性抗体通过Western blotting进行检测,并用eNOS和AS特异性抗体进行复制。印迹是三个独立实验的代表。NT公司,未处理。B类和D类,密度值分别表示从非处理对照组归一化为总eNOS或AS的变化百分比。密度变化显著,S.E.代表为误差线(n个= 3,第页< 0.05).E类,从用胰岛素或血管内皮生长因子处理过的BAEC培养基中提取的亚硝酸盐,采用DAN分析法,以S.E.表示误差线(*,n个= 3,第页< 0.05).
钙依赖性刺激BAEC增加Ser-328的磷酸化
胰岛素和VEGF通过PI3K/Akt途径介导Ser-1179处的eNOS磷酸化,该途径在很大程度上与钙无关(18–20). 由于胰岛素或VEGF介导的钙非依赖性信号传导促进了AS Ser-328的明显去磷酸化,因此我们质疑Ser-328处的磷酸化是否与钙依赖性信号传递有关。为了初步探讨这个问题,进行了定点突变分析。使用了三种结构体,一种包含野生型(WT)AS,一种含有Ser-328的Ala替代物(磷酸-全),另一种包含Ser-328(磷酸-甲基)的Asp替代物。如所示图3A类当用钙离子载体刺激时,磷酸型S328D AS的过度表达增强了NO的生成,类似于WT AS。相反,在相同的治疗中,过表达磷酸化全S328A AS未能提高NO的生成。Western blotting显示结构的一致表达(图3B类). 只有S328D过度表达将NO生成增加到WT水平的观察结果表明,Ser-328的磷酸化支持精氨酸在钙依赖性刺激条件下传递到eNOS。
图3。
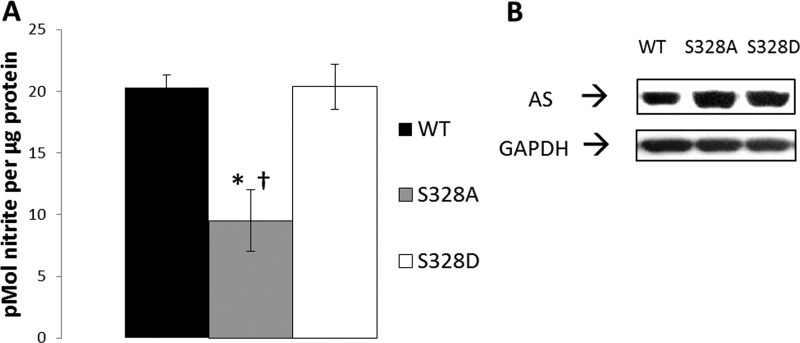
与WT或类磷酸化物(S328D)相比,过度表达的Ser-328磷酸化突变为丙氨酸产生的受刺激NO显著减少。
A类BAEC瞬时转染WT,将磷酸-全丝氨酸转染至丙氨酸328(S328A),或将磷酸-氨基酸丝氨酸转导至天冬氨酸(S328D)AS,然后用0.5μ米钙离子载体A23187和50μ米原钒酸钠。结果代表了S.E.的四个不同实验误差线. (*,第页相对于WT<0.05;†,第页<0.05(相对于S328D)。B类,代表性Western blot证实WT、S328A和S328D过度表达。
由于这些发现与钙依赖性一致,因此进行了实验,以确定Ser-328中的钙依赖性变化是否可用于内源性AS。在这些实验中,BAEC用钙离子载体A23187或内质网ATP酶抑制剂thapsigargin处理。众所周知,这两种药物都能促进细胞内钙的增加(21)刺激NO生成。如所示图4,A类和B类,两种药物均可促进eNOS在Ser-1179的磷酸化近50%,随后在Thr-497的eNOS磷酸化降低40%,与报告值一致(21,22). 然而,在这种情况下,用钙离子载体或thapsigargin治疗显示AS Ser-328的磷酸化增加(~60%)(图4,A类和B类).
图4。
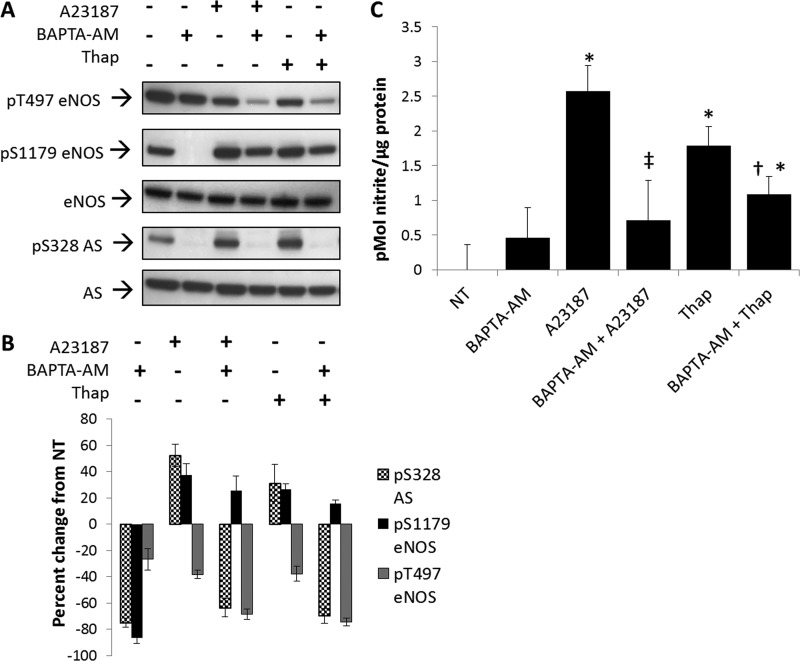
钙依赖性刺激NO生成增加精氨琥珀酸合成酶Ser-328的磷酸化。
A类磷酸化变化的Western blot分析(P(P))相对于用钙离子载体(A23187)和thapsigargin处理20分钟(十)在存在或不存在细胞内钙螯合剂BAPTA-AM的情况下。斑点是五个独立实验的代表。B类,密度值表示未经处理的(NT公司)对照分别归一化为总eNOS或AS。密度变化显著,S.E.代表为错误栏。C,在BAPTA-AM存在或不存在的情况下,用钙离子载体(A23187)和thapsigargin处理的BAEC培养基用DAN测定法测定亚硝酸盐。(n个=5,较未处理显著增加:*,第页<0.05,与A23187单独使用相比显著降低,第页<0.05,与单独使用thapsigargin相比显著降低;†,第页< 0.05).
为了进一步证实AS Ser-328磷酸化的钙依赖性,用细胞内钙螯合剂BAPTA-AM处理BAEC。正如预期的那样,钙离子载体和thapsigargin挽救了eNOS Ser(P)-1179处的磷酸化,而单独使用BAPTA-OM处理后磷酸化减弱。重要的是,如所示图4A类,用50μ处理米BAPTA-AM完全消除了Ser-328的磷酸化以及eNOS Ser-1179的磷酸化。因此,AS Ser-328的磷酸化似乎通过钙依赖信号与eNOS Ser-1179的磷酸化协调调节。
为了证明生理相关性,在用BAPTA-AM处理细胞30分钟,然后用钙离子载体A23187刺激20分钟后进行NO测量。A23187和thapsigargin与未处理或BAPTA-AM-处理的BAEC相比显著增加NO生成(图4C). 此外,BAPTA-AM处理显著降低了A23187或thapsigargin刺激的NO生成,分别为2.5倍和2倍。
PKC参与调节BAEC中Ser-328磷酸化
为了进一步阐明参与Ser-328处AS钙依赖性磷酸化的信号通路,使用激酶抑制剂进行了一系列实验。因为经典的PKC同型是钙依赖性激酶,不参与胰岛素或VEGF信号传导(23,24),我们假设PKC家族的一个成员可能参与AS Ser-328的钙依赖性磷酸化。为了验证这一假设,进行了非等型特异性PKC抑制研究。作为第一种方法,通过检测As Ser-328磷酸化直接测量PKC抑制的效果。如所示图5A类和B类与未经处理的对照组相比,用PKC抑制剂罗特列林或PKC抑制剂双吲哚甲基苯胺I治疗BAEC,然后用钙离子载体刺激,使AS Ser-328磷酸化降低了~60%。这些结果被认为是一致的,并且支持经典PKC同型在Ser-328促进AS磷酸化的作用。
图5。
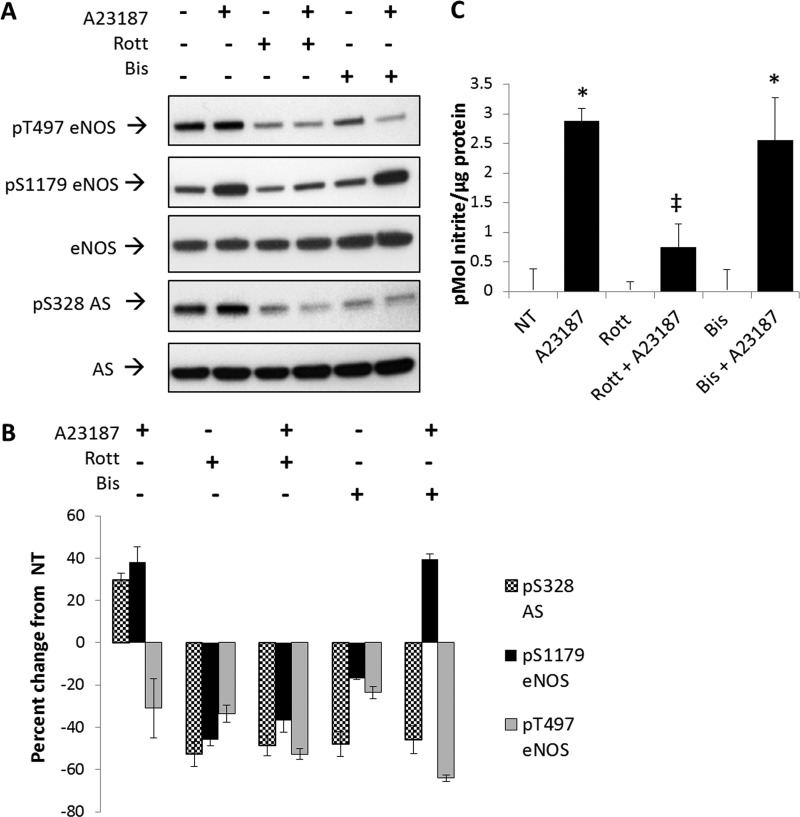
用42μ米罗特林或2.5μ米双吲哚甲酰胺I降低磷酸化(P(P))AS Ser-328。BAEC用42μ米鹿特林(Rott公司)或2.5μ米双吲哚马来酰亚胺I(比斯)在用钙离子载体(A23187)刺激前30分钟,持续20分钟。A类三个实验的代表性Western blot。B类,密度值表示未经处理的(NT公司)对照分别归一化为总eNOS或AS。密度变化显著,S.E.代表为误差线(n个= 3,第页< 0.05).C用DAN法检测处理后BAEC培养基中的亚硝酸盐。A23187在添加或不添加双吲哚甲酰胺I的情况下显著增加NO的生成(相对于未处理组的显著性:*,n个= 3,第页< 0.05; 相对于A23187的显著性:†,n个= 3,第页< 0.05).
有趣的是,无论钙刺激如何,经鹿特伦治疗后,eNOS的Thr-497或Ser-1179的磷酸化也降低了~60%。相反,当BAECS仅用双吲哚甲酰胺I治疗时,eNOS在Ser-1179和Thr-497的磷酸化仅下降20%。然而,在钙刺激下,eNOS Ser-1179的磷酸化增加到类似于单独钙离子载体治疗的水平。然而,在双吲哚甲酰胺I存在下,Ser-328的磷酸化不受钙的影响(图5A类).
为了证明系统对抑制剂反应的生理相关性,我们测量了每种治疗的NO生成(图5C). 用双吲哚甲酰胺I抑制PKC对BAEC产生NO的影响不大;然而,鹿特伦显著降低了钙离子载体刺激的NO生成2倍以上。这些结果表明,罗氏菌素可能影响几种激酶的活性,而双吲哚甲基苯胺I明显降低eNOS Thr-497的磷酸化,从而促进NO的生成,类似于单独钙离子载体。
为了进一步研究可能磷酸化Ser-328的其他钙依赖性激酶的潜在参与,进行了额外的激酶抑制研究。ERK1/2被PD98059抑制,Ca2+/用KN93抑制钙调素依赖性蛋白激酶II(CaMKII)。在这两种情况下,当BAEC与钙离子载体或thapsigargin联合处理时,未观察到对AS Ser-328磷酸化的抑制作用(数据未显示)。这些结果表明PKC,而不是ERK1/2或Ca2+/钙调素依赖性蛋白激酶II负责钙刺激的AS Ser-328磷酸化。
因为PKCα是一种钙依赖性激酶,在钙依赖性刺激eNOS期间被证明是活性的(25),我们试图确定这是否是负责AS Ser-328磷酸化的同型。为了测试这一点,我们检测了用DsiRNA直接敲低PKCα的效果。DsiRNA不同于用于基因沉默的传统siRNA,因为用于沉默的RNA双链不仅模仿骰子产物,但也通过RNA诱导沉默复合物(RISC)对骰子处理进行了优化,以产生更有效的击倒(26). 如所示图6,PKCα被显著击倒。针对PKCα的DsiRNA在1和5 n时允许50%和75%的敲除米DsiRNA。敲除PKCα后,用钙离子载体进行处理,发现1或5 n时Ser-328的磷酸化降低了50%和60%米并防止钙离子载体处理引起的磷酸化增加。在这些条件下,eNOS在Ser-1179的磷酸化在很大程度上不受PKCα敲除的影响,而eNOS Thr-497在PKCα被敲除80%时下降~60%。这些结果表明PKCα是一种磷酸化AS的Ser-328的激酶。
图6。
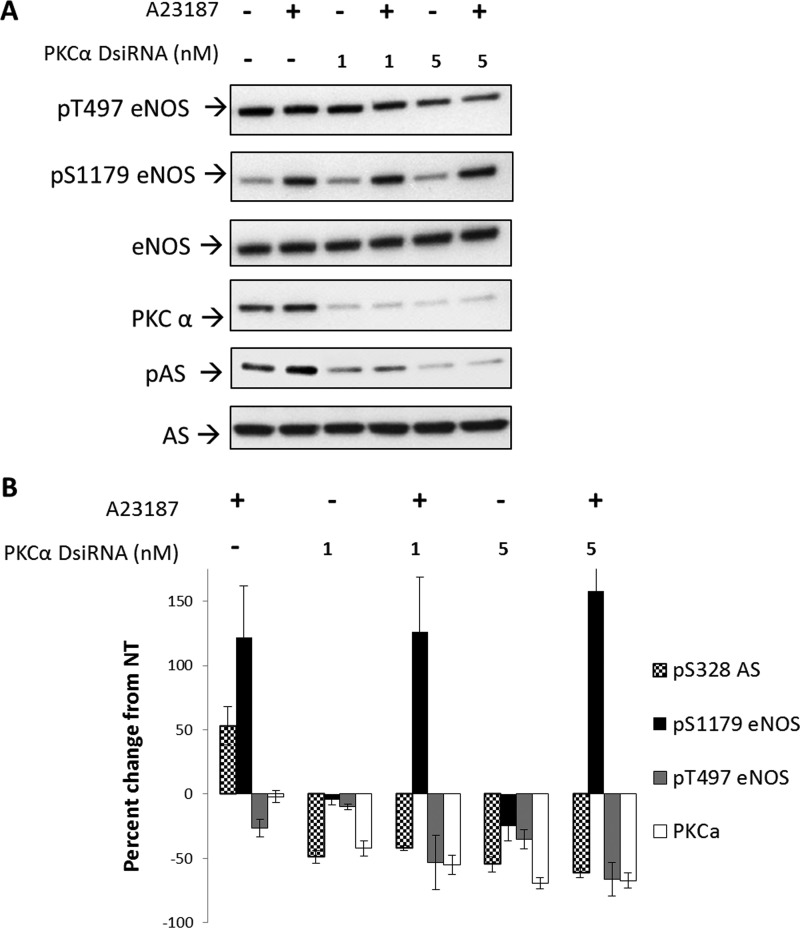
PKCα的敲除降低了磷酸化(P(P))位于Ser-328,而不是eNOS Ser-1179。BAEC用1或5 n米DsiRNA针对PKCα的第五外显子,并允许表达24小时,然后用钙离子载体A23187刺激20分钟,并与未处理的BAEC进行比较。A类三个实验的代表性Western blot。B类,密度值表示未经处理的(NT公司)对照分别归一化为总eNOS或AS。密度变化显著(除了A2317对PKCα表达或1 n米用S.E.表示的pS1179 eNOS上的DsiRNA处理误差线(n个= 3,第页<0.05)(相对于未处理或仅DsiRNA的显著性:*,n个= 3,第页< 0.05).
PKCα直接磷酸化物AS
由于观察到由于激酶敲除导致的磷酸化降低可能是信号级联中上游激酶抑制的结果在体外激酶分析用于确定PKCα是否直接磷酸化AS。基于缺乏一致的Akt磷酸化基序(RX(X)R(右)XX年(S/T))(27)在AS蛋白序列中。
如所示图7A类,PKCα清楚地证明了AS磷酸化的能力在体外。虽然Akt1空白反应的计数有所增加,但这低于被视为显著的阈值。SDS-PAGE分馏32P-标记的激酶反应产物和放射自显影胶片曝光的可视化证实PKCα磷酸化AS,而不是Akt1(图7B类).
图7。
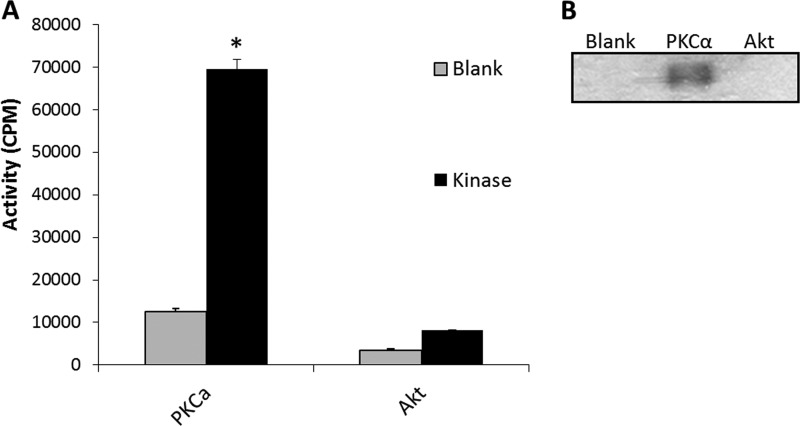
PKCα,但不是Akt,磷酸化AS在体外.
A类,在体外激酶分析。灰色条代表纯化的AS负激酶的培养(空白).黑色条表示纯化AS与指示激酶的孵育(激酶). 激酶活性用cpm表示误差线代表S.E(n个= 3, *,第页< 0.05).B类,代表性放射自显影在体外用SDS-PAGE分离的激酶反应如A类.
讨论
血管内皮一氧化氮的生成是一个动态的、有针对性的过程,其中eNOS的多级调节已经建立(28). 由于AS是NO生成所必需的,因此有理由推测AS的翻译后调节在维持NO稳态中发挥重要作用。事实上,最近使用质谱分析的工作表明,AS在Ser-352位点磷酸化(人类序列)(9); 然而,这种磷酸化的生物学意义尚未明确。此外,科尔宾等。(8)表明AS的磷酸化随着VEGF的治疗而改变,但磷酸化位点未知。在本研究中,我们证明AS Ser-328磷酸化支持钙依赖性刺激血管内皮细胞NO生成,并由PKCα介导。
AS参与催化瓜氨酸-NO循环中精氨酸再循环侧的速率限制步骤,对于维持内皮细胞中NO的生成至关重要(29). 与AS和eNOS之间已建立的转录共调控一致(30–32)本研究中所述的工作使用Western blot分析表明,在NO生成的刺激依赖于钙介导的信号传导的条件下,AS与eNOS在翻译后共同调节。进一步的研究表明,钙刺激过表达磷酸化物(S328D)AS的BAEC使NO生成增加到过表达WT AS的水平,而过表达的磷酸化物全(S328A)AS无法支持与WT对照相比NO生成的增加(图3). 此外,发现在基本条件下,空的和磷酸化的AS突变体结构都能够将NO的生成提高到野生型结构的水平,这也证实了两个过度表达的AS突突变体都具有代谢活性(数据未显示)。最后,通过激酶抑制剂研究和敲除分析,我们能够确定在钙介导的NO生成刺激下,PKCα促进了AS Ser-328的磷酸化。
考虑到AS在NO生成中的重要作用以及eNOS受钙水平调节的时空方式,AS易受钙变化调控机制的影响也就不足为奇了。事实上,VEGF或胰岛素对eNOS的钙非依赖性激活导致AS Ser-328的磷酸化降低,这一事实进一步将AS Ser-329的磷酸化与eNOS激活的钙依赖性途径区分开来。
此前,我们的实验室已经表明AS存在于eNOS的洞穴中(33). 因为PKCα在活化时与质膜相关(34),PKCα和AS之间的局部功能关系可能得到支持。此外,有报道称,细胞内钙的变化促进了eNOS的胞质再定位(5). 因此,我们也很容易推测,在钙介导的信号传导中,Ser-328处AS的磷酸化可能以与eNOS一致的方式促进细胞溶质AS的重新定位。这将确保精氨酸循环和易位eNOS之间的功能关系得以维持。目前正在进行实验来验证这个假设。
总之,这些结果首次证明了AS的生物学相关磷酸化位点,并与之前的研究结果一致,这些研究表明eNOS和AS相对于血管内皮NO生成的协调调节。鉴于AS在血管内皮NO生成中的关键作用,以及我们实验室以前的工作表明,AS的整体磷酸化随着VEGF治疗的改变而改变(8)这些结果还表明,其他磷酸化位点参与AS调控,这些位点值得进一步考虑。
致谢
我们感谢南佛罗里达大学化学系的Wayne Guida博士和Daniel Santiago博士在电子建模方面的专业知识,感谢H.Lee Moffitt癌症中心和研究所的John Kooman博士,感谢用于质谱分析的蛋白质组学核心设施,感谢Denise Cooper博士在PKC抑制方面的指导。
*这项工作得到了美国国立卫生研究院拨款R01 HL083153-01A2(致Duane C.Eichler)的全部或部分支持。这项工作还得到了佛罗里达州卫生部詹姆斯·埃斯特·金生物医学研究项目拨款(给杜安·埃希勒)的支持。
2使用的缩写如下:
- 电子NOS
内皮型一氧化氮合酶
- 作为
精氨琥珀酸合成酶
- BAEC公司
牛主动脉内皮细胞
- BAPTA-AM公司
1,2-二(o个-氨基苯氧基)乙烷-N个,N个,N个′,N个′-四乙酸(乙酰氧基甲酯)
- 丹
2,3-二氨基萘
- DsiRNA
骰子底物siRNA。
参考文献
-
1Flam B.R.、Eichler D.C.、Solomonson L.P.(2007)内皮一氧化氮的生成与瓜氨酸-NO循环紧密耦合。一氧化氮17, 115–121[内政部] [公共医学] [谷歌学者]
-
2Haines R.J.、Pendleton L.C.、Eichler D.C.(2011)精氨酸琥珀酸合成酶:精氨酸代谢的中心。国际生物化学杂志。分子生物学。2, 8–23[PMC免费文章] [公共医学] [谷歌学者]
-
三。Li H.,Meininger C.J.,Hawker J.R.,Jr.,Haynes T.E.,Kepka Lenhart D.,Mistry S.K.,Morris S.M.,Jr.,Wu G.(2001)精氨酸酶I和II在内皮细胞中一氧化氮、多胺和脯氨酸合成中的调节作用。美国生理学杂志。内分泌。Metab公司。280,E75–82[内政部] [公共医学] [谷歌学者]
-
4.Simon A.,Plies L.,Habermeier A.,MartinéU.,Reining M.,Closs E.I.(2003)中性氨基酸转运和蛋白质分解在人类内皮细胞一氧化氮合酶底物供应中的作用。循环。物件。93, 813–820[内政部] [公共医学] [谷歌学者]
-
5Prabhakar P.,Thatte H.S.,Goetz R.M.,Cho M.R.,Golan D.E.,Michel T.(1998)内皮一氧化氮合酶的受体调节易位。生物学杂志。化学。273, 27383–27388[内政部] [公共医学] [谷歌学者]
-
6Fleming I.(2010)eNOS激活的分子机制。Pflugers架构。459, 793–806[内政部] [公共医学] [谷歌学者]
-
7Michell B.J.、Chen Z.p.、Tiganis T.、Stapleton D.、Katsis F.、Power D.A.、Sim A.T.、Kemp B.E.(2001)通过蛋白激酶C和cAMP依赖性蛋白激酶协调控制内皮一氧化氮合酶磷酸化。生物学杂志。化学。276, 17625–17628[内政部] [公共医学] [谷歌学者]
-
8Corbin K.D.、Pendleton L.C.、Solomonson L.P.、Eichler D.C.(2008)精氨琥珀酸合成酶的蛋白激酶A生物化学磷酸化。生物物理学。Res.Commun公司。377, 1042–1046[内政部] [公共医学] [谷歌学者]
-
9Imami K.,Sugiyama N.,Kyono Y.,Tomita M.,Ishihama Y.(2008)使用煅烧的二氧化钛/C18双相柱,通过二维纳米LC-MS对培养的癌细胞进行磷酸蛋白质组自动分析。分析。科学。24, 161–166[内政部] [公共医学] [谷歌学者]
-
10Gospodarowicz D.、Moran J.、Braun D.、Birdell C.(1976)牛血管内皮细胞的克隆生长:作为生存剂的成纤维细胞生长因子。程序。国家。阿卡德。科学。美国。73, 4120–4124[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
11Pendleton L.C.、Goodwin B.L.、Flam B.R.、Solomonson L.P.、Eichler D.C.(2002)内皮精氨琥珀酸合成酶mRNA 5′非翻译区域多样性:组织特异性表达的基础设施。生物学杂志。化学。277, 25363–25369[内政部] [公共医学] [谷歌学者]
-
12Misko T.P.、Schilling R.J.、Salvemini D.、Moore W.M.、Currie M.G.(1993)生物样品中亚硝酸盐的荧光测定法。分析。生物化学。214, 11–16[内政部] [公共医学] [谷歌学者]
-
13.Brouet A.、Sonveaux P.、Dessy C.、Balligand J.L.、Feron O.(2001)Hsp90确保了从早期Ca2+-依赖于血管内皮生长因子暴露的内皮细胞中内皮一氧化氮合酶的晚期磷酸化依赖性激活。生物学杂志。化学。276, 32663–32669[内政部] [公共医学] [谷歌学者]
-
14Montagnani M.,Chen H.,Barr V.A.,Quon M.J.(2001)胰岛素刺激的eNOS激活与钙无关2+但需要Akt在Ser-1179进行磷酸化。生物学杂志。化学。276, 30392–30398[内政部] [公共医学] [谷歌学者]
-
15Kroll J.、Waltenberger J.(1999)血管内皮生长因子受体-2(KDR)的一种新功能:内皮细胞中对VEGF-A刺激的反应中一氧化氮的快速释放。生物化学。生物物理学。Res.Commun公司。265, 636–639[内政部] [公共医学] [谷歌学者]
-
16Montagnani M.、Ravichandran L.V.、Chen H.、Esposito D.L.、Quon M.J.(2002)胰岛素受体底物-1和磷脂酰肌醇依赖性激酶-1是内皮细胞中胰岛素刺激一氧化氮生成所必需的。摩尔内分泌学。16, 1931–1942[内政部] [公共医学] [谷歌学者]
-
17Pappetropoulos A.、GarcíA-CardeñA G.、Madri J.A.、Sessa W.C.(1997)一氧化氮的产生有助于人类内皮细胞中血管内皮生长因子的血管生成特性。临床杂志。投资。100, 3131–3139[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
18曾国,群明杰(1996),沃特曼抑制胰岛素刺激的一氧化氮生成:血管内皮细胞中的直接测量。临床杂志。投资。98, 894–898[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
19.Dimmeler S.、Dernbach E.、Zeiher A.M.(2000)血管内皮生长因子诱导的内皮细胞迁移需要Ser-1177处内皮一氧化氮合酶的磷酸化。FEBS信函。477, 258–262[内政部] [公共医学] [谷歌学者]
-
20Dimmeler S.、Fleming I.、Fisslthaler B.、Hermann C.、Busse R.、Zeiher A.M.(1999)通过Akt依赖性磷酸化激活内皮细胞中的一氧化氮合酶。自然399, 601–605[内政部] [公共医学] [谷歌学者]
-
21Xiao Z.,Wang T.,Qin H.,Huang C.,Feng Y.,Xia Y.(2011)内质网钙2+释放通过细胞外信号调节激酶(ERK)1/2介导的丝氨酸635磷酸化调节内皮一氧化氮合酶。生物学杂志。化学。286, 20100–20108[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
22Tran Q.K.,Watanabe H.(2006)内皮细胞中的钙信号。手。实验药理学。145–187[内政部] [公共医学] [谷歌学者]
-
23Rask-Madsen C.,King G.L.(2007),疾病机制:胰岛素抵抗和糖尿病中的内皮功能障碍。自然临床。实践。内分泌。Metab公司。3, 46–56[内政部] [公共医学] [谷歌学者]
-
24.Rask-Madsen C.,King G.L.(2008)内皮细胞中PKC-α和PKC-ϵ对VEGF信号的差异调节。动脉硬化。血栓。瓦斯克。生物。28, 919–924[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
25Partovian C.,Zhuang Z.,Moodie K.,Lin M.,Ouchi N.,Sessa W.C.,Walsh K.,Simons M.(2005)PKCα激活eNOS并增加动脉血流量体内.循环。物件。97, 482–487[内政部] [公共医学] [谷歌学者]
-
26Collingwood M.A.、Rose S.D.、Huang L.、Hillier C.、Amarzguioui M.、Wiiger M.T.、Soifer H.S.、Rossi J.J.、Behlke M.A.(2008)与高效骰子底物小干扰RNA相容的化学修饰模式。寡核苷酸18, 187–200[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
27Yaffe M.B.、Leparc G.G.、Lai J.、Obata T.、Volinia S.、Cantley L.C.(2001)基于基序的剖面扫描方法,用于全基因组信号通路预测。自然生物技术。19, 348–353[内政部] [公共医学] [谷歌学者]
-
28Dudzinski D.M.、Michel T.(2007)eNOS的生活史:伙伴和途径。心血管研究。75, 247–260[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
29Goodwin B.L.、Solomonson L.P.、Eichler D.C.(2004)精氨酸琥珀酸合成酶的表达是维持主动脉内皮细胞中一氧化氮生成和细胞存活所必需的。生物学杂志。化学。279, 18353–18360[内政部] [公共医学] [谷歌学者]
-
30Goodwin B.L.、Corbin K.D.、Pendleton L.C.、Levy M.M.、Solomonson L.P.、Eichler D.C.(2008)曲格列酮上调血管内皮精氨琥珀酸合成酶。生物化学。生物物理学。Res.Commun公司。370, 254–258[内政部] [公共医学] [谷歌学者]
-
31Goodwin B.L.、Pendleton L.C.、Levy M.M.、Solomonson L.P.、Eichler D.C.(2007)肿瘤坏死因子-α降低主动脉内皮细胞中精氨琥珀酸合成酶的表达和一氧化氮的产生。美国生理学杂志。心脏循环。生理学。293,H1115–H1121[内政部] [公共医学] [谷歌学者]
-
32Haines R.J.、Corbin K.D.、Pendleton L.C.、Meininger C.J.、Eichler D.C.(2012)胰岛素转录调节精氨琥珀酸合成酶以维持血管内皮功能。生物化学。生物物理学。Res.Commun公司。421, 9–14[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
33.Flam B.R.、Hartmann P.J.、Harrell-Booth M.、Solomonson L.P.、Eichler D.C.(2001)精氨酸再生酶、精氨琥珀酸合成酶和裂解酶与内皮一氧化氮合成酶的小泡定位。一氧化氮5, 187–197[内政部] [公共医学] [谷歌学者]
-
34Rosse C.、Linch M.、Kermorgant S.、Cameron A.J.、Boeckeler K.、Parker P.J.(2010)《PKC与局部信号动力学控制》。自然修订版分子细胞生物学。11, 103–112[内政部] [公共医学] [谷歌学者]

