本文阐述了NF-κB在癌症治疗中的相反作用,并强调了癌症途径的上下文依赖性。使用跨物种方法,比较人类和小鼠模型系统,并将数据外推到患者队列,Jing等人确定了致癌网络,其中化疗激活的NF-κB信号不再介导耐药性,而是促进衰老,并有助于癌症治疗的结果。这与Chien等人的文章有关。
关键词:癌症治疗、细胞衰老、淋巴瘤、小鼠模型、NF-κB、DLBCL
摘要
在恶性肿瘤中,核因子-κB(NF-κB)活性增强在很大程度上被视为一种致癌特性,也会对化疗产生耐药性。最近,NF-κB被假定参与衰老相关和可能的衰老增强细胞因子反应,从而表明NF-κ的抑瘤作用。使用小鼠淋巴瘤模型并分析淋巴瘤患者的转录组和临床数据,我们发现治疗诱导的衰老表现为并依赖于活性NF-κB信号传导,而NFκB同时促进对凋亡的抵抗。根据不同的NF-κB相关致癌网络对原发性小鼠淋巴瘤进行进一步表征和基因工程,使我们想起弥漫性大B细胞淋巴瘤(DLBCL)亚型,这指导我们确定Bcl2-过表达生发中心B细胞样(GCB)DLBCL作为一个临床相关亚组,在NF-κB过度激活时具有显著优越的结果。我们的数据说明了跨物种研究在转基因小鼠肿瘤中功能测试遗传机制的能力,这些转基因小鼠肿瘤再现了相应人类实体的不同特征,并最终使用小鼠模型衍生的遗传信息重新定义新的临床相关患者亚群。
核因子κB(NF-κB)转录因子最初是通过与免疫球蛋白增强子序列的相互作用而确定的,它控制着一大组靶基因的表达,从而协调了多种生物功能,包括细胞生存、炎症和免疫(森和巴尔的摩1986;海登和戈什2008). 具体来说,与癌症相关的特性,如抑制凋亡、促进生长、增强迁移和侵袭性,都归因于NF-κB信号(Ben-Neriah和Karin 2011). NF-κB转录因子或其上游信号成分的激活突变在各种淋巴瘤实体中常见(Lenz等人,2008a;Compagno等人,2009年; 有关审查,另请参阅Staudt 2010年以及其中的参考)。虽然NF-κB信号介质中的致癌突变在实体肿瘤中似乎很少见,但在没有突变的情况下,致癌信号可能会增强NF-κ的活性,因为炎症相关NF-kb B驱动的细胞因子的产生以非细胞自主的方式促进致癌(Staudt 2010年;Ben-Neriah和Karin 2011). 因此,小鼠模型显示NF-κB在淋巴瘤和实体瘤的发展中具有致癌作用(Basseres等人,2010年;Calado等人,2010年;Yang等人,2010年). 然而,其他基于小鼠模型的研究发现,NF-κB转录因子或上游激活物并非作为肿瘤抑制因子(Luedde等人,2007年;Keller等人,2010年)从而强调了NF-κB网络介导效应器功能的复杂性和潜在的上下文依赖性(Perkins和Gilmore 2006年). 事实上,NF-κB已被证明在不同的环境中介导或相反地阻断细胞凋亡(Wang等人,1998年;Ryan等人,2000年)而NF-κB作用的非细胞自主成分可能进一步增加了NFκB在肿瘤发展中明显发挥的多种作用。
NF-κB的激活也发生在对DNA损伤的反应中(Brach等人,1991年;Stilmann等人,2009年;Hinz等人,2010年)大多数常规化疗药物发挥抗癌活性的方式。在抑制NF-κB功能后观察到增强的抗肿瘤疗效,并主要与NF-κ)B介导的凋亡抑制有关,这是化疗耐药的潜在机制(Wang等人,1999年). 考虑到NF-κB调控的生物反应的复杂性,它们对肿瘤发展的双重影响,以及致癌基因和化疗药物对其中许多反应的诱导性,除抗细胞凋亡外,NF-κB的效应功能可能会导致治疗效果受损(Li和Sethi 2010)然而,并非所有与药物相关的NF-κB控制功能都会降低化疗敏感性。
细胞早衰是一种通过致癌激活或抗癌化疗诱发的DNA损伤反应(DDR)信号而急性启动的终末细胞周期阻滞程序,作为一种应激诱导的失效安全机制,与凋亡互补(施密特2007;Kuilman等人,2010年). 虽然癌基因诱导衰老(OIS)是针对Ras/Raf型癌基因进行的最深入研究,但它在早期(前)恶性病变中施加了抗肿瘤屏障(Serrano等人,1997年;Braig等人,2005年),治疗性衰老(TIS)已被证明可以改善癌症治疗的长期结果(Schmitt等人,2002年). 重要的是,OIS和TIS都表现出衰老相关分泌表型(SASP)(Coppe等人,2008年),一系列主要促炎细胞因子,据报道,这些细胞因子的自分泌或旁分泌作用至少在某些情况下会加强衰老的阻滞,它们与天然宿主免疫系统的相互作用可能解释了免疫介导的衰老肿瘤细胞的清除(Xue等人,2007年;Acosta等人,2008年;Kuilman等人,2008年). 许多SASP因子是真正的NF-κB靶点,其启动子中有NF-κ的B结合位点(Feuerhake等人,2005年;Acosta等人,2008年;Kuilman等人2008)如聚(ADP-核糖)聚合酶-1(PARP-1)介导的基因毒性治疗或致癌Ras诱导(Finco等人,1997年;Stilmann等人,2009年;Ohanna等人,2011年). 衰老的人成纤维细胞中NF-κB转录因子的沉默增强了其增殖能力,这表明NFκB信号可能确实有助于延缓衰老生长(Roneank等人,2011年).
在这项研究中,我们使用了初级Eμ-myc公司转基因小鼠淋巴瘤是人类侵袭性B细胞非霍奇金淋巴瘤(B-NHL)的成熟模型,以及来自人类弥漫性大B细胞淋巴瘤(DLBCL,最常见的侵袭性B-NHL类型)的信息采用跨物种方法确定致癌网络,其中化疗激活的NF-κB信号不再介导耐药性,而是促进细胞衰老,并有助于抗癌治疗的结果。
结果
TIS中NF-κB活性增加
我们之前建立了淋巴蛋白Eμ-myc公司转基因小鼠作为TIS模型系统(Schmitt等人,2002年;Reimann等人,2007年). 分离原发性真菌驱动的B细胞淋巴瘤(以下称为“对照”淋巴瘤),进行逆转录病毒治疗β细胞淋巴瘤/白血病基因2转导阻断细胞凋亡,并暴露于DNA损伤的化疗药物阿霉素(ADR)。第5天,ADR治疗对照组;bcl2淋巴瘤完全生长停滞,增殖标志物Ki67染色阴性,并均匀地表现出衰老相关的β-半乳糖苷酶活性(SA-β-gal)(补充图S1A,B;Dimri等人,1995年). 为了揭示转录组水平上TIS相关的变化,我们从12对匹配的主要对照组中生成了全基因组微阵列基因表达谱(GEP);药物衰老或未经治疗的bcl2淋巴瘤。基因集富集分析(GSEA)表明,NF-κB靶基因作为一个整体,特别是NF-κ的B控制的细胞因子,强烈倾向于TIS组(图1A; 补充表1)。支持这一结果,一项基于多重DNA-结合ELISA的未经治疗与衰老淋巴瘤配对分析检测到衰老细胞中NF-κB家族四个亚单位p50、p52、p65(RelA)和RelB的DNA-绑定活性显著升高,表明TIS中经典和替代NF-κB通路均被激活(图1B). 因此,TIS淋巴瘤中免疫荧光染色的p65主要位于细胞核,而非基因细胞显示出优先的细胞质定位(图1C). 通过免疫印迹分析,TIS淋巴瘤还表现出Ser 536磷酸化p65(p65-P-S536)的诱导表达水平,这种翻译后修饰被认为可以增加p65反式激活功能(图1D;Buss等人,2004年). 为了确认单个靶基因水平的NF-κB活性,我们通过定量实时PCR(RQ-PCR)测量了微阵列分析中差异调节基因产物中转录物的表达。事实上,与非基因淋巴瘤细胞相比,所有NF-κB靶基因检测非分泌基因和SASP在TIS中的表达水平更高(图1E). 总之,活化的NF-κB信号传导是化疗相关肿瘤细胞衰老的一个显著特征。
图1。
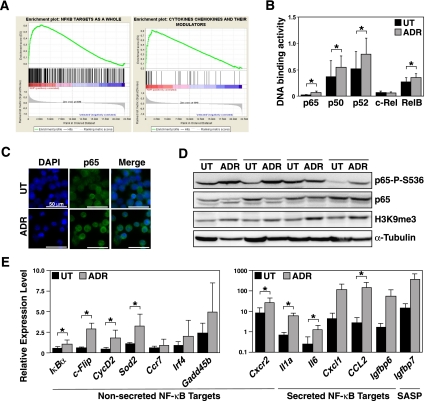
治疗诱导的衰老原发性淋巴瘤细胞NF-κB活性增强。(A类)NF-κB靶点的GSEA(左边)细胞因子及其调节剂的子集(正确的)ADR衰老与未治疗(ut)Eμ的GEP-myc公司控制;bcl2淋巴瘤(n个=12对匹配的配对)。(B类)淋巴瘤中指定NF-κB亚单位的DNA结合活性A类(n个每亚单位≥3对配对)。(C类)淋巴瘤细胞胞浆制备中NF-κB亚单位p65的免疫荧光A类(分析了三个案例的代表性示例;bars,50μm)。(D类)四对匹配淋巴瘤细胞裂解液中指示蛋白的免疫印迹分析A类以H3K9me3作为衰老标记(Reimann等人,2010年)α-管蛋白作为负荷控制。(E类)RQ-PCR分析显示的非分泌型(左边)和秘密的(正确的)淋巴瘤中的转录物A类(n个=每个样品5个)。所示为相对表达式水平,标准化为内部控件,在所有数据集中都是可比较的。(注意细胞因子面板中的对数标度显示。此处显示的所有分泌NF-κB靶点反映SASP,而免疫球蛋白7是SASP因子,但不是真正的NF-κB靶点。)所有柱状图条表示平均值±标准偏差(SDEV)。
TIS依赖体内完整的NF-κB功能
接下来,我们询问TIS是否不仅表现出增强的NF-κB活性,而且取决于它。首先,我们测试了NF-κB超级阻遏物IκBα-ΔN(SR)-一种不可降解的IκBα部分的稳定表达是否使NF-κB失活(Krappmann等人,1996年)肿瘤坏死因子-α(TNF-α)治疗后NF-κB靶基因表达水平降低,对未治疗淋巴瘤细胞的增殖影响很小(补充图S2A,B)-足以干扰TIS条件的功能特性。事实上,在ADR暴露的SR工程对照组中,大多数NF-κB靶基因(SASP和非分泌基因)的表达显著降低;bcl2淋巴瘤组,在对具有或不具有代表性NF-κB控制的SASP因子SR部分的相同单个淋巴瘤的配对分析中进一步强调(图2A; 有关暴露于多种药理性IKK抑制剂后获得的类似结果,请参见补充图S2C)。基于这些发现,我们提出了一个明显的问题,即NF-κB活性的消融是否会影响化疗后衰老的诱导性。事实上,对仅在SR状态上不同的配对进行的基于SA-β-gal的分析显示,从一些SA-β-gal阳性细胞的百分比大幅下降到SR队列中其他淋巴瘤的未受损TIS表型,与空载体组相比,SR中SA-β-gal反应性平均降低了30%以上,相应的细胞数平均增加了2.5倍以上(图2B; 数据未显示)。总的来说,这些数据表明,无论是细胞内、自分泌还是旁分泌的作用方式,在同型肿瘤细胞群中,推测的前发性NF-κB调控功能的作用不大(Acosta等人,2008年;Kuilman等人,2008年)-至少在一部分遗传易感原发性肿瘤中。NF-κB网络不是作为DDR诱导衰老途径的一个线性和基本组成部分发挥作用,可以很好地想象,它是前发性DDR的一个证实性的、间接的中继。在这种情况下,所有细胞在培养基中均匀暴露于化疗药物可能会在每个给定细胞中启动如此强大的DDR级联反应,以致SR对衰老增强原理的定量减少不会立即转化为衰老缺陷表型,但侧支前发途径会出现功能障碍,从而使NF-κB信号对TIS的贡献至关重要。
图2。
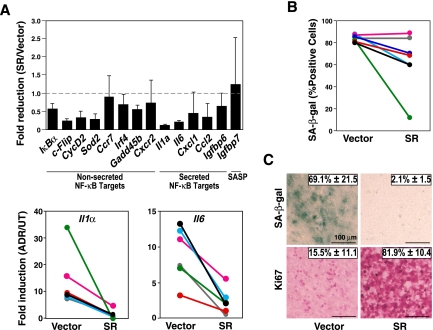
TIS是原发性淋巴瘤中的NF-κB依赖性疾病。(A类,顶部)淋巴瘤中指示转录物的RQ-PCR分析,以ADR暴露与未治疗对照的相对值之比表示;稳定表达NF-κB SR或被空载体感染的bcl2淋巴瘤作为对照(每个至少5个样本)。(底部)匹配ADR暴露对照;bcl2淋巴瘤对±SR显示为两个代表性SASP因子的相对表达水平。(B类)体外暴露于ADR的匹配淋巴瘤对±SR中SA-β-gal阳性细胞的频率(n个=7对)。(C类)体内暴露于CTX的配对(±SR)淋巴瘤切片中SA-β-gal阳性和Ki67阳性细胞的原位频率(n个=3对;所示的代表性显微照片;棒,100μm)。所有柱状图条或数字表示平均值±SDEV。
体内情况要复杂得多,包括不均匀药物输送和体外分析未涵盖的异型细胞-细胞相互作用等方面。为了解决NF-κB活性对药物诱导衰老表型的潜在贡献,我们决定研究体内遗传相容性移植淋巴瘤。控制;将逆转录病毒感染SR构建物或空载体的bcl2淋巴瘤移植到正常、免疫活性的受体小鼠中,在那里它们形成的系统性淋巴瘤与它们最初来源的原始转基因宿主没有区别。在可触及外周淋巴结病时,对具有类似肿瘤负担的小鼠进行一次DNA损伤抗癌药物环磷酰胺(CTX)治疗,并在5天后分析淋巴瘤的TIS原位征象。有趣的是,体内CTX暴露后,TIS-活性淋巴瘤几乎没有SA-β-gal反应性,如果表达SR部分,则Ki67阳性细胞>80%(图2C). 因此,SR表达的淋巴瘤在体内表现出更为深刻的TIS缺陷,即使它们在体外仅表现出SA-β-半乳糖反应性的一些降低,这表明旁观者细胞在肿瘤微环境中具有关键的NF-κB依赖性促生作用。值得注意的是,正如Lowe及其同事报道的那样,衰老与宿主免疫细胞的相关相互作用也受到NF-κB活性的共同控制(Chien等人,2011年; 另请参阅Xue等人,2007年)因此,提示免疫介导的衰老细胞清除是NF-κB控制肿瘤生长的另一个环节。综上所述,NF-κB网络是药物诱导衰老反应的关键组成部分,因此其对细胞自主和非细胞自主衰老相关过程的干扰的实际定量影响只能在体内进行评估。
致癌网络决定NF-κB在治疗结果中的相反作用
鉴于SR工程化的分枝淋巴瘤在体内无法启动TIS反应,我们旨在量化和分析NF-κB信号对自然形成的B细胞淋巴瘤在任何治疗之前的长期预后的贡献。虽然在候选途径中具有明确遗传损伤的癌症小鼠模型是重要的研究工具,但它们可能会错过甚至干扰驱动自然发展肿瘤的关键致癌网络相互作用。因此,我们从一系列12个初级Eμ-myc公司淋巴瘤,通过p65 DNA-结合ELISA评估其内源性NF-κB激活水平,并将其分为“NF-κ子B低”(NL,最低的四例)和“NF-kb高”(NH,最高的四例(图3A). 首先,我们试图确定其短期药物敏感性,以反映其凋亡潜力。预期NH组实际上对体外ADR治疗产生了耐药性,而NL组的淋巴瘤细胞在相同剂量水平下定量死亡(图3B). 由于Bcl2在人类B细胞淋巴瘤和真正的NF-κB靶点中经常过度表达,我们推断NH淋巴瘤中可能通过Bcl2的诱导介导化疗耐药性,至少部分是这样的(Schmitt等人,2000年;Feuerhake等人,2005年). 的确,Bcl2公司通过RQ-PCR测量,NH组的转录水平显著高于NL组(图3C). 当我们从两组逆转录病毒等位基因中过度表达Bcl2时,Bcl2使NL淋巴瘤与NH淋巴瘤一样具有化疗耐药性(图3D). 相反,我们询问抑制NF-κB活性是否可以克服NH组的短期耐药性。虽然SR部分对NF-κB功能的失活在NL组中几乎没有影响,但它恢复了NH组的化疗敏感性,从而揭示了在化疗期间作为该组“致命弱点”的高活性NFκB信号(图3E;Staudt 2010年). 综上所述,一部分原发性Myc-driven淋巴瘤在诊断时表现出高水平的NF-κB活性,这表明在淋巴瘤发展过程中选择的NF-kb B网络具有致癌功能,同时预测了原发性耐药性。
图3。
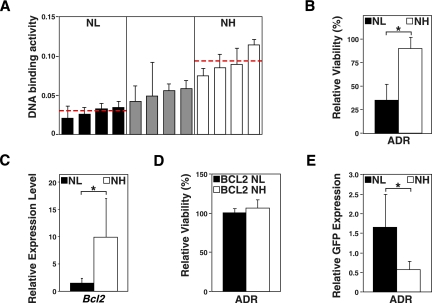
Eμ-myc公司由高内源性NF-κB水平驱动的淋巴瘤显示出NFκB/Bcl2介导的化疗耐药性。(A类)根据诊断时的NF-κB p65 DNA结合活性(测量三次;水平线表示各组内的平均活性),将12个主要对照淋巴瘤分层,分为“NFκB低”(NL;黑条)、“中间”(灰条)和“NF-κ的B高”(NH;白条)组。(B类)通过台盼蓝染料排除NL和NH淋巴瘤细胞群在体外暴露24小时至5纳克/毫升ADR相对于同一组未处理细胞的活性分析(n个=每组4人)。(C类)NF-κB靶点转录水平的RQ-PCR分析β细胞淋巴瘤/白血病基因2淋巴瘤组如B类. (D类)可行性分析,如B类在相同的淋巴瘤样本中,两组的逆转录病毒等位基因Bcl2均过度表达。(E类)MSCV-SR-IRES-GFP表达细胞的GFP富集分析B类,但持续48小时;NH组GFP阳性细胞的选择性下降表明其对NF-κB抑制的化疗敏感性增加(n个=每组3人)。所有柱状图条表示平均值±SDEV。
总结DLBCL患者的临床结果,~60%的小鼠携带Eμ-myc公司对照组淋巴瘤获得长期缓解(反映治愈),而其余40%在CTX诱导治疗后复发(Schmitt等人,1999年,2000;Lenz等人,2008b). 我们生成了13个原发性(非β细胞淋巴瘤/白血病基因2-感染)Eμ-myc公司淋巴瘤移植到受体小鼠体内,并在淋巴瘤出现时暴露于CTX。当所有动物都进入缓解期时,有7例淋巴瘤在100天的观察期内复发,我们可以将诊断时的13例淋巴瘤分为“永不复发”(NR)或“复发预防”(RP)。基于先前的结果,我们通过表达IκBα,一个中央NF-κB调节器和NF-κ)B靶点,以及Bcl2公司在这两组未经治疗的淋巴瘤中(图4A). 引人注目的是,RP淋巴瘤的发病率明显高于IκBα表达水平进一步升高β细胞淋巴瘤/白血病基因2表达水平,从而使人想起NF-κB/Bcl2网络在图3在活化B细胞样(ABC)DLBCL中可检测到的遗传标志性病变中,预测性较低的DLBCL亚型的特征是NF-κB通路中的多种激活突变导致组成性活性NF-κ的B信号传导,并且具有非常高的Bcl2公司转录水平(Iqbal等人,2006年;Lenz等人,2008b;Compagno等人,2009年;Staudt 2010年;Nogai等人,2011年).
图4。
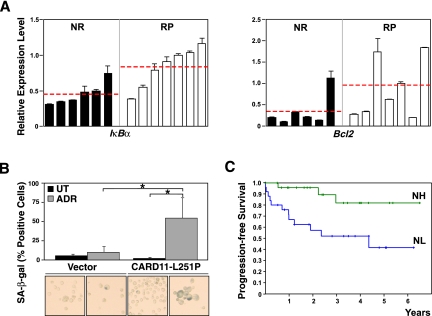
不同的致癌连线决定了NF-κB在治疗结果中的相反作用。(A类)根据体内移植瘤形成后对CTX治疗的长期反应将13个主要对照淋巴瘤分为“永不复发”(NR)或“复发型”(RP)淋巴瘤,并对其进行RQ-PCR分析IκBα诊断时(即在任何治疗之前)的转录水平作为NF-κB靶点的代表(左边)和,共Bcl2公司相同样品的水平(正确的; 以相同顺序呈现);水平线表示各组内的平均活动。(B类)Bcl2-过度表达NL淋巴瘤±CARD11-L251P中SA-β-gal阳性细胞的频率,体外暴露3 d至50 ng/mL ADR(n个=4对配对;显示了胞浆制剂的代表性显微照片在下面相应的条)。(C类)共233名患者中49名R-CHOP样治疗的GCB DLBCL患者的无进展生存率Lenz等人2008年b)中点以上Bcl2公司诊断时通过基于微阵列的转录组分析对其淋巴瘤进行表达,并通过63基因特征进一步分层为“NF-κB高”(NH,n个= 25; 绿色)或“NF-κB低”(NL,n个= 24; 蓝色)。所有柱状图条表示平均值±SDEV。
相反,生发中心B细胞样(GCB)DLBCL具有更好的临床结果(Lenz等人,2008b)尽管Bcl2过表达在该亚型中也很常见。重要的是,GCB DLBCL很少具有活化的NF-κB突变,但经常(~45%)在t(14;18)易位的背景下发展,该易位以NF-κB非依赖性的方式通过免疫球蛋白重链基因座驱动Bcl2过表达,这种病变在ABC型DLBCL中几乎从未发现(Nogai等人,2011年). 如果外源性Bcl2过度表达阻断小鼠淋巴瘤中药物诱导的凋亡,从而模拟GCB相关t(14;18)易位,则NR淋巴瘤能够进入TIS(数据未显示)。为了测试更高的NF-κB激活水平是否会进一步促进这些淋巴瘤的衰老诱导,我们使用了一种强烈的NFκB活化卡片11(也称为CARMA1公司或Bimp3型)突变体,人类CARD11-L244P突变体的小鼠同源物,其过度表达导致NF-κB靶基因表达增加(补充图S3;Lenz等人,2008a). 自然发生时卡片11突变优先出现在ABC DLBCL中,我们在这里使用这种突变来增强“GCB回忆”淋巴瘤队列中的NF-κB活性,该队列是在缺乏高度激活的NF-kb B的情况下形成的,并且凋亡通过自身Bcl2过度表达而独立于NF-kbB被阻断。事实上,在早期,当ADR暴露的Bcl2-engineed NL淋巴瘤在TIS症状方面仍基本呈阴性时,高比例的匹配突变CARD11-过度表达淋巴瘤细胞已被SA-β-gal活性染色为阳性(图4B)表明较高的NF-κB活性通过TIS促进治疗结果。
因此,我们决定对233名患者进行利妥昔单抗-CHOP样标准免疫化疗(包括ADR和CTX)后的DLBCL GEP和临床数据进行大数据集查询(Lenz等人,2008b),并在Bcl2高表达的GCB DLBCL患者子集中,询问由63个已知NF-κB靶基因组成的先前确定的NF-κ的B基因表达特征是否允许我们将两组临床结果不同的患者进行分层(Shaffer等人,2006年). 的确,如所示图4C,Bcl2公司-NF-κB标志性表达高于中位数(NH;n个=25)与补充NL组相比,无进展生存率显著提高(n个= 24;P(P)<0.005),而在携带高Bcl2公司-表达ABC亚型淋巴瘤或低表达Bcl2公司表达式(补充图S4;未显示数据)。值得注意的是,多变量分析表明NF-κB状态Bcl2公司-过度表达的GCB DLBCL病例在统计上与国际预后指数总分无关(P(P)=0.3956),用作预测淋巴瘤患者预后的临床标准(1993年国际非霍奇金淋巴瘤预测因素项目). 总之,我们的实验策略确定了一个遗传定义的、具有临床意义的GCB DLBCL患者亚组,如果NF-κB信号传导过度活跃,这些患者的治疗效果会更好,从而对比了ABC亚型与组成性活性NF-κB信号传导和不良治疗结果的既定关联。
讨论
细胞衰老不仅作为肿瘤抑制原理,而且有助于体内癌症治疗的结果(Schmitt等人,2002年;Haugsetter等人,2010年). 活化的NF-κB信号主要与治疗抵抗有关,尤其是在短期试验中,当被测量为无法进入药物诱导的凋亡时(Wang等人,1999年). 先前的研究表明,OIS与大量产生多种细胞因子有关,其中许多细胞因子在其启动子中具有NF-κB结合位点,我们在此报告了TIS期间非分泌和分泌NF-κ子B靶基因的显著诱导,重要的是,衰老表型对功能性NF-κB信号的依赖性在活体中变得尤为明显——这一发现与Lowe及其同事在本期的相关工作完全一致(Chien等人,2011年). 虽然由于突变的Card11表达而强制NF-κB活化在体外加速了TIS,但与体外情况相比,体内NF-κ子B抑制后TIS的损伤更为明显,这表明NF-κ的B信号在衰老过程中具有额外的非细胞自主影响。我们观察到,在NF-κB感受性淋巴瘤大量衰老时,NF-κB缺陷淋巴瘤细胞几乎没有衰老,这让人想起最近报道的巨噬细胞在Eμ-myc公司淋巴腺病(Reimann等人,2010年).
虽然我们的结果表明,在基因定义的模型场景中,NF-κB在诱导细胞衰老中发挥着重要作用,但NF-κ的活性水平对自然肿瘤治疗结果的总体影响更难评估。我们的跨物种调查策略有助于揭示具有明显NF-κB依赖性反应性的基因定义肿瘤亚群。基于人类DLBCL数据,将ABC亚型与激活NF-κB损伤联系起来,并且与标准免疫化疗的结果较差(Lenz等人,2008b;Compagno等人,2009年),我们首先将一系列初级Eμ-myc公司根据表现时NF-κB活性水平,体内对化疗具有已知长期反应的转基因淋巴瘤。回忆GCB型DLBCL,Eμ组-myc公司临床预后良好的淋巴瘤主要由基础NF-κB活性低的淋巴瘤组成。随后,这些小鼠淋巴瘤被用于实验研究逆转录病毒Bcl2过表达独立阻断药物诱导的凋亡后,NF-κB活性增强对衰老诱导的贡献。最终,我们使用了TIS易发场景GCB减少淋巴瘤的遗传决定因素Bcl2公司表达和高NF-κB活性,以研究233例临床疗效已知的DLBCL患者的大型GEP集。令人惊讶的是,我们能够在DLBCL宿主患者中证实小鼠淋巴瘤模型的机制性结果。“Bcl2公司治疗后生存期显著延长的患者的高/NF-κB高“GCB-DLBCL亚组强调了淋巴瘤患者中NF-κB过度激活与治疗结果之间迄今未知的有益联系,如果没有对原发性Eμ-myc公司淋巴瘤。我们的方法说明了小鼠模型在重述人类肿瘤队列中定义的遗传决定因素方面的能力,随后对小鼠替代肿瘤进行基因操作和随后的功能探索,最终目标是使用小鼠衍生的机制信息来重新评估人类数据集,以获得新的功能见解。
我们的数据强调了肿瘤发展过程中连接起来的致癌网络和相互依赖性如何调节NF-κB的实际功能,甚至在随后的治疗反应中的相反作用。正如在DLBCL的ABC亚型中优先观察到的那样,淋巴瘤发展过程中获得的NF-κB突变,解释了这些淋巴瘤对NFκB抑制剂或靶向NF-kb B途径成分的shRNAs的选择性脆弱性(Davis等人,2001年;Ngo等人,2006年). 自从抗凋亡β细胞淋巴瘤/白血病基因2基因是一个真正的NF-κB靶点,ABC DLBCL在基因上决定依赖NF-κ的B/Bcl2信号模块,而GCB DLBCL经常与β细胞淋巴瘤/白血病基因2将其抗凋亡活性与NF-κB上游对照解耦的易位。与肿瘤不同的是,肿瘤具有广泛的有害生物学特性,这些生物学特性与构成活性NF-κB网络直接相关,那些不依赖NF-κB信号传导作为致癌驱动力,但存在NFκB非依赖性凋亡阻滞的肿瘤类型,可能利用NF-κ子B信号传导的原发潜能来执行TIS,作为一种替代的、结果改善的化疗效应器机制。我们在设计为过度表达Bcl2的Myc-driven淋巴瘤中获得的小鼠模型发现,不仅在基因上接近人类Myc/Bcl2“双重命中”淋巴瘤(最常见于DLBCL的GCB亚型),而且还包括更晚期的滤泡性淋巴瘤,除了其t(14;18)标志性病变外,通常在疾病过程中获得激活的Myc病变,值得注意的是,也不属于NF-κB过度激活的淋巴瘤实体(Aukema等人,2011年). 尽管核因子-κB在癌症发展和治疗中的作用非常复杂,但我们的小鼠模型和患者衍生数据表明,我们应该仔细选择或至少监测基因定义的患者亚群,在这些亚群中,对核因子-kb B通路的治疗干预可能会或可能不会有临床益处(Dunleavy等人,2009年;Ruan等人,2011年).
材料和方法
小鼠、淋巴瘤监测和体内治疗
本研究中使用的所有动物方案均经政府审查委员会(柏林Landesamt Berlin)批准,并符合各自的监管标准。Eμ-myc公司通过等位基因特异性基因组PCR对转基因小鼠进行基因分型,并监测淋巴瘤发病情况(Adams等人,1985年;Schmitt等人,1999年). 分离活的淋巴瘤细胞,通过尾静脉注射将其移植到应变匹配的C57BL/6受体小鼠(~3×106至5×106每只小鼠的细胞数),或在培养基中繁殖,并按照所述进行snap冷冻或福尔马林固定淋巴结组织的保存(Schmitt等人,1999年). 在受体动物可触及淋巴结的那一刻,将小鼠暴露于单次腹腔内剂量的CTX(300 mg/kg体重),并通过淋巴结触诊或治疗后第5天分离的淋巴瘤材料来监测反应(Schmitt等人,1999年,2002).
定点突变、质粒和逆转录病毒基因转移
致癌CARD11-L251P突变(在ABC DLBCL细胞系OCL-Ly3中检测到的人类CARD11-L244P突变的小鼠同源物)(Lenz等人,2008a)由全长野生型小鼠CARD11(BioCat)的定点突变产生。编码小鼠野生型CARD11、CARD11-L251P、Bcl2和NF-κB SR的cDNA(Krappmann等人,1996年)被亚克隆到小鼠干细胞逆转录病毒(MSCV;即,进入MSCV-IRES-GFP或MSCV主干中,同时存在一个短杆菌素或嘌呤霉素抗生素耐药基因)。用带有相应MSCV质粒的Phoenix包装细胞瞬时转染产生的逆转录病毒上清液稳定感染Eμ-myc公司转基因淋巴瘤细胞(Schmitt等人,2000年).
生长参数和体外药物测定
通过台盼蓝染料排除法评估细胞数量和细胞活性,并在添加指定浓度的ADR后24 h或5 d分别测量细胞毒性或细胞衰老(Schmitt等人,1999年;Reimann等人,2010年). 在一些实验中,淋巴瘤细胞预先暴露于NF-κB抑制剂Bay11-7082(1μM;IκBα磷酸化抑制剂;Sigma-Aldrich)、TPCA-1(0.5μM;IKK-2抑制剂;Merck)或KINK-1(0.4μM;一种IKK-1抑制剂;由M.Schmidt-Supprian善意提供[Schon等人,2008年])在随后的ADR治疗之前,或在50μM浓度下用TNF-α(Sigma-Aldrich)治疗30分钟或24小时。对于MSCV-SR-IRES-GFP感染细胞的GFP富集分析,ADR暴露前后GFP阳性细胞百分比的变化通过用MSCV-GFP对照载体感染的GFP阳性细胞的各个部分进行标准化。如前所述,在snap冰冻淋巴结冰冻切片中进行悬浮培养的细胞分裂素制备,用于随后的抗原或SA-β-半乳糖活性检测染色以及原位SA-β-gal分析(Schmitt等人,2002年;Braig等人,2005年;Reimann等人,2010年).
NF-κB DNA结合活性
根据制造商的方案,使用TransAm Flexi NFκB家族转录因子ELISA分析(Active Motif)测量五种哺乳动物NF-κB亚单位的DNA结合活性。
基因表达分析
为了对淋巴瘤细胞中NF-κB靶基因进行RQ-PCR分析,使用SuperScript逆转录酶(Invitrogen)和随机六聚体或寡核苷酸将用TRIzol(Invit罗gen)提取的RNA转录到cDNA中。小鼠RQ-PCR分析Bcl2公司,CCL2级,CCR7号机组,c-FLIP公司,CXCL1系列,CXCR2型,周期素D2抗原,GADD45b公司,GAPDH公司,GM-CSF公司,IGFBP6型,IGFBP7型,IκBα,IL-1α,白细胞介素-6,IRF-4型、和SOD2标准转录本是基于商用引物(Applied Biosystems)进行的。对于每个给定的样本,ΔCt值被确定为特定转录物的Ct值与GAPDH公司作为内务管理控制mRNA,然后根据2(-ΔΔCt)ΔΔCt=ΔCt治疗−ΔCt未经处理的.
使用在十二烷基硫酸钠(SDS)样品缓冲液(60 mM Tris-HCl,pH 6.8,10%甘油,2%SDS和5%2-巯基乙醇)中裂解细胞生成的全细胞裂解物进行免疫印迹,在12%SDS-PAGE上进行解析,并使用Bcl2抗体转移到Immobilon-P膜(Millipore)(1:2000稀释;#554087,BD Pharmingen)(数据未显示),H3K9me3(1:1000;ab8898,Abcam),NF-κB p65(1:1000,#3034,细胞信号技术),NFκB P 65-P-S536(1:1000:#3033,细胞信号技术),以及α-管蛋白(1:2500;T5168,Sigma)作为负载控制(Schmitt等人,1999年). 对于免疫荧光,细胞固定在4%多聚甲醛中,用0.1%Triton X-100/PBS渗透,用1%牛血清白蛋白封闭,用抗p65的一级抗体(1:250;Santa Cruz Biotechnologies,SC-372)孵育,然后用0.01%吐温20作为洗涤剂缓冲液和二级抗兔IgG抗体(1:400;AlexaFluor 488[A11008],Invitrogen);用DAPI(4′,6-二氨基-2-苯基吲哚)对载玻片进行染色,作为核复染,并用Mowiol 4-88(钙生物化学)固定载玻片。为了对Ki67进行免疫染色,将2%的副甲醛固定胞浆制剂或组织冷冻切片与抗Ki67的一级抗体(1:25;M7240,Dako)孵育,然后使用链霉亲和素-生物素复合物过氧化物酶试剂盒(LSAB+System-AP,DakoCytomation),根据制造商的说明使用。
对于基于微阵列的基因表达谱,从未处理或5-d ADR处理的对照中分离RNA;bcl2 Eμ-myc公司使用RNeasy minikit(Qiagen)的淋巴瘤,并根据制造商的说明与Affymetrix小鼠基因组430 2.0微阵列(Affymmetrix)杂交。阵列在GeneChip Fluidics Station 450中进行杂交和清洗,并在Affymetrix GeneChip3000扫描仪上检测到信号。Affymetrix文件被导入Partek Genomic Suite软件(6.4版,Partek,Inc.),并由实现的稳健多阵列(RMA)工作流程进行处理(包括中值抛光探针集摘要、RMA背景校正和分位数归一化)。原始微阵列数据以登录号存放在国家生物技术信息中心的基因表达综合(GEO)存储库GSE31099标准(网址:http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31099).
为了进一步分析小鼠微阵列数据,在没有进一步改变的情况下,从Thomas Gilmore实验室网站获取了NF-κB激活的基因签名(完整的基因列表和相关参考文献可在http://www.bu.edu/nf-kb/gene-resources/target-genes). NF-κB靶基因集在未经治疗与ADR治疗对照组配对的全球转录信号中的富集;应用GSEA 2.0版软件(麻省理工大学布罗德学院和哈佛大学,http://www.broad.mit.edu/gsea). 归一化富集分数(NES)反映了P(P)-值<0.05,FDR(错误发现率)值<0.25(Subramanian等人,2005年).
233名接受R-CHOP样免疫化疗并使用Affymetrix HG-U133 Plus 2.0微阵列进行分析的患者的淋巴瘤活检的基因表达数据(Lenz等人,2008b),来自NCBI GEO(GSE10846标准). 患者按照以下标准分为ABC DLBCL、GCB DLBCL和非分类DLBCLLenz等人(2008b)100例GCB DLBCL患者样本(可获得无进展生存期的临床随访信息)均分为高、低两组Bcl2公司表达式,基于中值表达式。在这两个队列中,患者随后根据之前描述的由63个已知NF-κB靶基因组成的NFκB基因表达特征的中位数表达分为高NF-κ子B组和低NF-κ子B组(http://lymphochip.nih.gov/cgi-bin/signaturedb/signature DB_DisplayGenes.cgi?signatureID=83;Shaffer等人,2006年).
统计评估
使用Kaplan-Meier估计计算GEP相关患者亚群的生存率,并与log-rank(Mantel-Cox)检验进行比较。皮尔逊χ2-多元分析中采用了检验法。未成婚者吨-试验用于比较平均值和标准偏差。染色反应的所有定量(即免疫染色或SA-β-gal分析)均由独立且盲法的第二位检查者进行,并反映至少三个样本,每个样本至少统计200个事件(通常在三个以上不同区域)。A类P(P)-值<0.05被认为具有统计学意义,并用星号标记。
致谢
我们感谢M.Schmidt-Supbrian提供的试剂;N.Burbach、A.Herrmann、B.Teichmann和S.Wegener提供技术援助;和施密特实验室的成员进行讨论和编辑建议。这项工作得到了德国Forschungsgemeinschaft(TRR 54)对M.H.、B.D.、G.L.、C.S.、C.A.S.和S.L.的资助;德意志联邦银行向G.L.和C.A.S.提供;来自慈善实验和临床研究中心和Max-Delbrück-Center for Molecular Medicine的B.D.、C.S.和C.A.S;以及Berliner Krebsgesellschaft e.V.和Else Kröner-Fresenius-Stiftung的G.L。
工具书类
-
Acosta JC、O’Loghlen A、Banito A、Guijarro MV、Augert A、Raguz S、Fumagalli M、Da Costa M、Brown C、Popov N等,2008年。通过CXCR2受体的趋化因子信号传导增强衰老。手机133:1006–1018[内政部] [公共医学] [谷歌学者]
-
Adams JM、Harris AW、Pinkert CA、Corcoran LM、Alexander WS、Cory S、Palmiter RD、Brinster RL 1985。c-myc公司免疫球蛋白增强子驱动的癌基因在转基因小鼠中诱导淋巴恶性肿瘤。自然318:533–538[内政部] [公共医学] [谷歌学者]
-
Aukema SM、Siebert R、Schuuring E、van Imhoff GW、Kluin-Nelemans HC、Boerma EJ、Kruin PM 2011。双位B细胞淋巴瘤。血液117:2319–2331[内政部] [公共医学] [谷歌学者]
-
Basseres DS、Ebbs A、Levantini E、Baldwin AS 2010。NF-κB亚单位p65/RelA在K-Ras诱导的肺肿瘤发生中的需求。癌症研究70:3537–3546[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Ben Neriah Y,Karin M 2011年。炎症与癌症相遇,NF-κB是媒人。自然免疫12:715–723[内政部] [公共医学] [谷歌学者]
-
Brach MA、Hass R、Sherman ML、Gunji H、Weichselbaum R、Kufe D 1991年。电离辐射诱导核因子κB的表达和结合活性。临床投资杂志88:691–695[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Braig M、Lee S、Loddenkemper C、Rudolph C、Peters AH、Schlegelberger B、Stein H、Dorken B、Jenuwein T、Schmitt CA 2005。癌基因诱导的衰老是淋巴瘤发展的初始屏障。性质436:660-665[内政部] [公共医学] [谷歌学者]
-
Buss H、Dorrie A、Schmitz ML、Hoffmann E、Resch K、Kracht M,2004年。丝氨酸536处p65 NF-κB的组成性和白细胞介素1诱导的磷酸化由多种蛋白激酶介导,包括IκB激酶(IKK)-α、IKKβ、IKKɛ、TRAF家族成员相关(TANK)-结合激酶1(TBK1)、,和一种未知激酶,将p65与TATA-结合蛋白相关因子II31介导的白细胞介素-8转录偶联。生物化学杂志279:55633–55643[内政部] [公共医学] [谷歌学者]
-
Calado DP、Zhang B、Srinivasan L、Sasaki Y、Seagal J、Unitt C、Rodig S、Kutok J、Tarakhovsky A、Schmidt-Supprian M等,2010年。在活化的B细胞样弥漫性大细胞淋巴瘤的发病机制中,典型NF-κB活化与BLIMP1的破坏协同作用。癌细胞18:580–589[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Chien Y、Scuoppo C、Wang X、Fang X、Balgley B、Bolden JE、Premsrirut P、Luo W、Chicas A、Lee CS等,2011年。NF-kB对衰老相关分泌表型的控制促进衰老并增强化疗敏感性。基因开发(本期)doi:10.1101/gad.17276711[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Compagno M、Lim WK、Grunn A、Nandula SV、Brahmachary M、Shen Q、Bertoni F、Ponzoni M、Scandura M、Califano A等人,2009年。多个基因的突变导致弥漫性大B细胞淋巴瘤中NF-κB的失调。自然459:717–721[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Coppe JP、Patil CK、Rodier F、Sun Y、Munoz DP、Goldstein J、Nelson PS、Desprez PY、Campisi J 2008。衰老相关分泌表型揭示了致癌RAS和p53抑癌基因的细胞自主功能。《公共科学图书馆·生物》6:2853–2868[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Davis RE、Brown KD、Siebenlist U、Staudt LM,2001年。活化的B细胞样弥漫性大B细胞淋巴瘤细胞的生存需要组成核因子κB活性。《实验医学杂志》194:1861–1874[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Dimri GP、Lee X、Basile G、Acosta M、Scott G、Roskelley C、Medrano EE、Linskens M、Rubelj I、Pereira-Smith O等人,1995年。一种生物标记物,用于识别培养中和体内老化皮肤中的衰老人类细胞。国家科学院院刊92:9363–9367[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Dunleavy K、Pittaluga S、Czuczman MS、Dave SS、Wright G、Grant N、Shovlin M、Jaffe ES、Janik JE、Staudt LM等人,2009年。硼替佐米联合化疗对弥漫性大B细胞淋巴瘤分子亚型的疗效差异。血液113:6069–6076[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Feuerhake F、Kutok JL、Monti S、Chen W、LaCasce AS、Cattoretti G、Kurtin P、Pinkus GS、de Leval L、Harris NL等,2005年。原发性纵隔大B细胞淋巴瘤和弥漫大B细胞恶性淋巴瘤亚型的NFκB活性、功能和靶基因特征。血液106:1392–1399[内政部] [公共医学] [谷歌学者]
-
Finco TS、Westwick JK、Norris JL、Beg AA、Der CJ、Baldwin AS Jr 1997年。致癌Ha-Ras诱导的信号传导激活NF-κB转录活性,这是细胞转化所必需的。生物化学杂志272:24113–24116[内政部] [公共医学] [谷歌学者]
-
Haugsetter AM、Loddenkemper C、Lenze D、Grone J、Standfuss C、Petersen I、Dorken B、Schmitt CA,2010年。细胞衰老预测转移性结直肠癌的治疗结果。英国癌症杂志103:505–509[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
海登理学硕士,Ghosh S 2008。NF-κB信号传导的共同原理。手机132:344–362[内政部] [公共医学] [谷歌学者]
-
Hinz M、Stilmann M、Arslan SC、Khanna KK、Dittmar G、Scheidereit C,2010年。细胞质ATM–TRAF6–cIAP1模块将核DNA损伤信号与泛素介导的NF-κB活化联系起来。分子细胞40:63–74[内政部] [公共医学] [谷歌学者]
-
1993年国际非霍奇金淋巴瘤预测因素项目。侵袭性非霍奇金淋巴瘤的预测模型。《新英格兰医学杂志》329:987–994[内政部] [公共医学] [谷歌学者]
-
Iqbal J、Neppalli VT、Wright G、Dave BJ、Horsman DE、Rosenwald A、Lynch J、Hans CP、Weisenburger DD、Greiner TC等,2006年。BCL2表达是活化的B细胞样弥漫性大B细胞淋巴瘤的预后标志。临床肿瘤学杂志24:961–968[内政部] [公共医学] [谷歌学者]
-
Keller U、Huber J、Nilsson JA、Fallahi M、Hall MA、Peschel C、Cleveland JL,2010年。Myc对Nfkb2的抑制会加速淋巴腺病。BMC癌症10:348 doi:10.1186/1471-2407-10-348[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Krappmann D,Wulczyn FG,Scheidereit C,1996年。不同的机制控制体内NF-κB抑制剂IκBα的信号诱导降解和基础转换。EMBO期刊15:6716–6726[PMC免费文章] [公共医学] [谷歌学者]
-
Kuilman T、Michallou C、Vredeveld LC、Douma S、van Doorn R、Desmet CJ、Aarden LA、Mooi WJ、Peeper DS 2008。癌基因诱导的衰老通过白细胞介素依赖性炎症网络传递。细胞133:1019–1031[内政部] [公共医学] [谷歌学者]
-
Kuilman T、Michallou C、Mooi WJ、Peeper DS 2010。衰老的本质。基因开发24:2463–2479[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Lenz G、Davis RE、Ngo VN、Lam L、George TC、Wright GW、Dave SS、Zhao H、Xu W、Rosenwald A等人,2008a。人类弥漫性大B细胞淋巴瘤的癌基因CARD11突变。科学319:1676-1679[内政部] [公共医学] [谷歌学者]
-
Lenz G、Wright G、Dave SS、Xiao W、Powell J、Zhao H、Xu W、Tan B、Goldschmidt N、Iqbal J等,2008b。大B细胞淋巴瘤的基质基因特征。《新英格兰医学杂志》359:2313–2323[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Li F,Sethi G,2010年。靶向转录因子NF-κB以克服癌症治疗中的化疗耐药和放疗耐药。Biochim生物物理学报1805:167-180[内政部] [公共医学] [谷歌学者]
-
Luedde T、Beraza N、Kotsikoris V、van Loo G、Nenci A、De Vos R、Roskams T、Trautwein C、Pasparakis M,2007年。肝实质细胞中NEMO/IKKγ的缺失会导致脂肪性肝炎和肝细胞癌。癌细胞11:119–132[内政部] [公共医学] [谷歌学者]
-
Ngo VN、Davis RE、Lamy L、Yu X、Zhao H、Lenz G、Lam LT、Dave S、Yang L、Powell J等人,2006年。癌症分子靶点的功能丧失RNA干扰筛选。性质441:106–110[内政部] [公共医学] [谷歌学者]
-
Nogai H、Dorken B、Lenz G,2011年。非霍奇金淋巴瘤的发病机制。临床肿瘤学杂志29:1803–1811[内政部] [公共医学] [谷歌学者]
-
Ohanna M、Giuliano S、Bonet C、Imbert V、Hofman V、Zangari J、Bille K、Robert C、Bressac-de Paillerets B、Hofman-P等人,2011年。衰老细胞产生PARP-1和核因子-κB相关分泌体(PNAS)。基因Dev 25:1245–1261[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Perkins ND,Gilmore TD 2006年。好警察,坏警察:NF-κB的不同面孔。细胞死亡差异13:759–772[内政部] [公共医学] [谷歌学者]
-
Reimann M、Loddenkemper C、Rudolph C、Schildhauer I、Teichmann B、Stein H、Schlegelberger B、Dorken B、Schmitt CA,2007年。Myc-voked DNA损伤反应解释了体内原发性淋巴瘤的治疗耐药性。血液110:2996–3004[内政部] [公共医学] [谷歌学者]
-
Reimann M、Lee S、Loddenkemper C、Drr JR、Tabor V、Aichele P、Stein H、Dörken B、Jenuwein T、Schmitt CA,2010年。肿瘤基质衍生的TGF-β通过Suv39h1依赖性衰老限制了真菌驱动的淋巴腺病。癌细胞17:262–272[内政部] [公共医学] [谷歌学者]
-
2011年,罗反派E、曼斯菲尔德L、卡埃塔诺C、阿尔瓦雷斯·费尔南德斯M、卡巴列罗OL、梅德玛RH、胡默里奇H、贾特PS。核因子-κB信号的激活促进细胞衰老。癌基因30:2356–2366[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
阮J、Martin P、Furman RR、Lee SM、Cheung K、Vose JM、Lacasce A、Morrison J、Elstrom R、Ely S等,2011年。硼替佐米联合CHOP-利妥昔单抗治疗先前未经治疗的弥漫性大B细胞淋巴瘤和套细胞淋巴瘤。临床肿瘤学杂志29:690–697[内政部] [公共医学] [谷歌学者]
-
Ryan KM、Ernst MK、Rice NR、Vousden KH 2000。NF-κB在p53介导的程序性细胞死亡中的作用。自然404:892–897[内政部] [公共医学] [谷歌学者]
-
Schmitt CA 2007年。细胞衰老与癌症治疗。Biochim生物物理学报1775:5–20[内政部] [公共医学] [谷歌学者]
-
Schmitt CA、McCurrach ME、de Stanchina E、Wallace-Brodeur RR、Lowe SW,1999年。INK4a/ARF突变通过抑制p53而加速淋巴腺病并促进化疗抵抗。基因Dev 13:2670–2677[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Schmitt CA,罗森塔尔CT,Lowe SW 2000。原发性小鼠淋巴瘤化疗耐药性的遗传分析。国家医学6:1029–1035[内政部] [公共医学] [谷歌学者]
-
Schmitt CA、Fridman JS、Yang M、Lee S、Baranov E、Hoffman RM、Lowe SW 2002。由p53和p16INK4a控制的衰老程序有助于癌症治疗的结果。手机109:335–346[内政部] [公共医学] [谷歌学者]
-
Schon M、Wienrich BG、Kneitz S、Sennefelder H、Amschler K、Vohringer V、Weber O、Stiewe T、Ziegelbauer K、Schon MP 2008。KINK-1,一种新型IKKβ小分子抑制剂,以及黑色素瘤细胞对抗肿瘤治疗的敏感性。美国国家癌症研究所杂志100:862–875[内政部] [公共医学] [谷歌学者]
-
Sen R,巴尔的摩D,1986年。多个核因子与免疫球蛋白增强子序列相互作用。手机46:705–716[内政部] [公共医学] [谷歌学者]
-
Serrano M,Lin AW,McCurrach ME,Beach D,Lowe SW 1997年。癌基因ras引起与p53和p16INK4a积累相关的细胞过早衰老。手机88:593–602[内政部] [公共医学] [谷歌学者]
-
Shaffer AL、Wright G、Yang L、Powell J、Ngo V、Lamy L、Lam LT、Davis RE、Staudt LM,2006年。阐明正常和病理淋巴生物学的基因表达特征库。免疫学修订版210:67–85[内政部] [公共医学] [谷歌学者]
-
Staudt LM 2010年。NF-κB的致癌激活。冷泉Harb Perspect Biol 2:a000109 doi:10.1101/cshperspect.a000109[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Stilmann M、Hinz M、Arslan SC、Zimmer A、Schreiber V、Scheidereit C,2009年。核多聚(ADP-核糖)依赖性信号体提供DNA损伤诱导的IκB激酶激活。摩尔细胞36:365–378[内政部] [公共医学] [谷歌学者]
-
Subramanian A、Tamayo P、Mootha VK、Mukherjee S、Ebert BL、Gillette MA、Paulovich A、Pomeroy SL、Golub TR、Lander ES等,2005年。基因集富集分析:解释全基因组表达谱的基于知识的方法。国家科学院院刊102:15545–15550[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Wang CY、Mayo MW、Korneluk RG、Goeddel DV、Baldwin AS Jr,1998年。NF-κB抗凋亡:诱导TRAF1和TRAF2以及c-IAP1和c-IAP2抑制caspase-8激活。科学281:1680–1683[内政部] [公共医学] [谷歌学者]
-
Wang CY,Cusack JC Jr,Liu R,Baldwin AS Jr,1999年。诱导性耐药的控制:通过抑制NF-κB增加凋亡来增强抗肿瘤治疗。国家医学5:412–417[内政部] [公共医学] [谷歌学者]
-
Xue W、Zender L、Miething C、Dickins RA、Hernando E、Krizhanovsky V、Cordon-Cardo C、Lowe SW 2007。衰老和肿瘤清除是由小鼠肝癌中p53的恢复触发的。自然445:656–660[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Yang J、Splittgerber R、Yull FE、Kantrow S、Ayers GD、Karin M、Richmond A,2010年。Ikkb的有条件消融抑制小鼠黑色素瘤的发展。临床研究杂志120:2563–2574[内政部] [PMC免费文章] [公共医学] [谷歌学者]

