

摘要
介绍
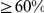
材料和方法
1.基准数据集
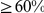
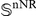
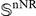
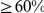
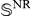
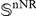
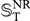
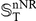
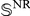
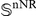
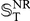
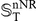
表1。 学习数据集的细分 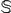
和测试数据集 
.
|
||||
|
|
|||
|
||||
|
||||
|
||||
|
||||
|
||||
|
||||
|
||||
|
||||
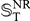
|
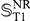
|
|||
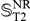
|
||||
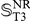
|
||||
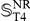
|
||||
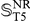
|
||||

|
||||

|
||||
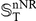
|
||||
2.序列衍生特征
2.1氨基酸组成(AAC)
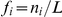
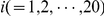
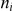

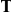
2.2二肽组成(DC)
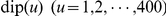
 |
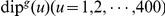
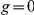
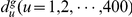
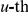
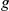
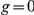
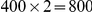
图1。 显示沿着蛋白质链具有不同间隙的二肽的示意图。
![]()
2.3复杂性因素(CF)
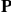
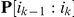
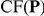
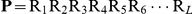
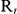
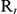
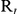
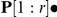
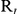
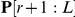
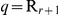
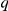
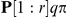
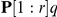
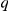
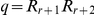

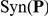
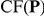
2.4傅里叶频谱成分(FSC)
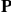
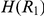
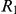
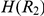
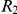
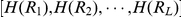
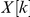
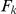
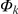
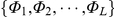
2.5特征融入伪氨基酸成分(PseAAC)
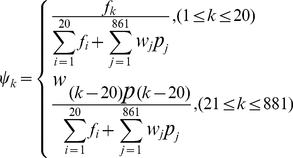 |
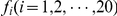
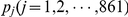
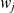
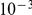
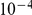
2.6模糊K最近邻(FKNN)分类器
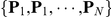
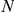
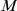
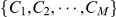
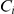
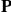
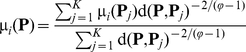 |
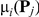
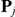
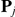
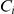
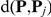
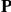
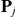
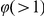
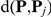
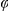
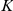
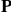


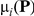
图2。 显示NR-2L操作过程的流程图。
![]()
结果和讨论
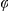
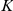
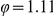
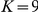
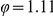
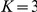
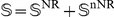
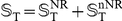
图3。 3D图形显示不同参数的折刀成功率。
![]()
表2。 通过折刀试验和独立数据集试验预测NR和非NR识别的成功率和MCC指数。
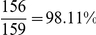
|
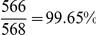
|
|||
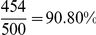
|
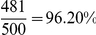
|
|||
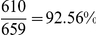
|
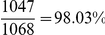
|
|||
表3。 通过jackknife检验和独立检验鉴定NR亚家族的预测成功率和MCC指数。
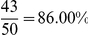
|
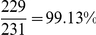
|
|||
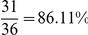
|
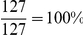
|
|||
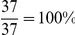
|
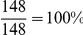
|
|||
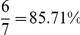
|
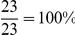
|
|||
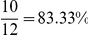
|
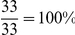
|
|||
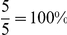
|
||||
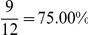
|
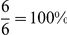
|
|||
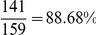
|
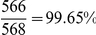
|
|||
图4。 预测结果在四个象限中的分布。
![]()
表4。 通过分别使用基准数据集上的不同特征,在识别NR亚家族时获得的折刀成功率 支持信息S1 .
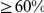
表5。 识别具有不同冗余减少截止阈值的NR亚家族时获得的折弯成功率 一 .
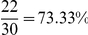
|
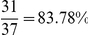
|
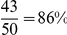
|
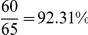
|
|
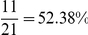
|
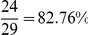
|
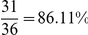
|
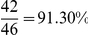
|
|
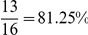
|
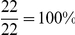
|
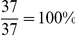
|
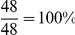
|
|
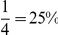
|
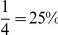
|
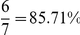
|
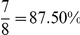
|
|
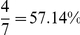
|
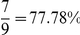
|
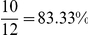
|
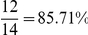
|
|
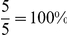
|
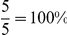
|
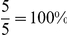
|
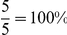
|
|
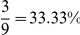
|
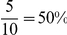
|
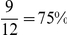
|
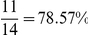
|
|
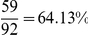
|
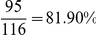
|
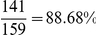
|
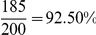
|
支持信息
致谢
脚注
工具书类
-
1 Altucci L,Gronemeyer H。细胞生与死中的核受体。 内分泌与代谢趋势。 2001; 12:460–468. doi:10.1016/s1043-2760(01)00502-1。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
2 Mangelsdorf DJ、Thummel C、Beato M、Herrlich P、Schutz G等。核受体超家族:第二个十年。 单元格。 1995年; 83:835–839. doi:10.1016/0092-8674(95)90199-x。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ] -
三。 Robinson-Rechavi M,Garcia HE,Laudet V.核受体超家族。 细胞科学杂志。 2003; 116:585–586. doi:10.1242/jcs.00247。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
4 Florence H、Gerrit V、Fred EC。收集和收集生物数据:GPCRDB和NucleaRDB信息系统。 核酸研究。 2001; 29:346–349. doi:10.1093/nar/29.1.346。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ] -
5 Altschul SF、Madden TL、Schäffer AA、Zhang J、ZhangZ等。缺口BLAST和PSI-BLAST:新一代蛋白质数据库搜索程序。 核酸研究1997; 25:3389–3402. doi:10.1093/nar/25.17.3389。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ] -
6 周家川。 使用伪氨基酸成分预测蛋白质细胞属性。 蛋白质。 2001; 43:246–255. doi:10.1002/port.1035。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
7 Bhasin M,Raghava GPS。 基于氨基酸组成和二肽组成的核受体分类。 生物化学杂志。 2004; 279:23262–23266. doi:10.1074/jbc。 M401932200。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
8 高庆斌,金振聪,叶XF,吴C,何J.用最佳伪氨基酸组成预测核受体。 分析生物化学。 2009; 387:54–59. doi:10.1016/j.ab.2009.01.018。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
9 周家川。 关于蛋白质属性预测和伪氨基酸组成的一些评论(50周年回顾)。 理论生物学杂志。 2011; 273:236–247. doi:10.1016/j.jtbi.2010.12.24。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ] -
10 Li W,Godzik A.Cd-hit:一个快速程序,用于聚类和比较大组蛋白质或核苷酸序列。 生物信息学。 2006; 22:1658–1659. doi:10.1093/bioinformatics/btl158。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
11 周家川。 一种预测(20–1)-D氨基酸组成空间中蛋白质结构类的新方法。 蛋白质:结构、功能和生物信息学。 1995年; 21:319–344. doi:10.1002/port.340210406。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
12 Nakashima H,Nishikawa K,Ooi T。蛋白质的折叠类型与氨基酸组成有关。 生物化学杂志。 1986; 99:153–162. doi:10.1093/oxfordjournals.jbchem.a135454。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
13 周GP。关于蛋白质结构类预测的有趣争议。 蛋白质化学杂志。 1998; 17:729–738. doi:10.1023/a:1020713915365。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
14 周GP,K博士。凋亡蛋白的亚细胞定位预测。 蛋白质:结构、功能和遗传学。 2003; 50:44–48. doi:10.1002/port.10251。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
15 刘伟,周朝中。 蛋白质二级结构含量预测。 蛋白质工程,1999; 12:1041–1050. doi:10.1093/protein/12.12.1041。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
16 周家川。 用于预测蛋白质中HIV蛋白酶裂解位点的矢量化序列耦合模型。 生物化学杂志。 1993; 268:16938–16948. [ 公共医学 ] [ 谷歌学者 ] -
17 周家川。 综述:预测蛋白质中的急转弯及其类型。 分析生物化学。 2000; 286:1–16. doi:10.1006/abio.2000.4757。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
18 肖X,邵SH,黄ZD,周KC。 用伪氨基酸组成预测蛋白质结构类别:用复杂性度量因子探讨。 计算化学杂志。 2006; 27:478–482。 doi:10.1002/jcc.20354。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
19 Gusev VD,Nemytikova LA,Chuzhanova NA。关于遗传序列的复杂性度量。 生物信息学。 1999; 15:994–999. doi:10.1093/bioinformatics/15.12.994。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
20 周家川。 综述:生物大分子的低频集体运动及其生物功能。 生物物理化学。 1988; 30:3–48. doi:10.1016/0301-4622(88)85002-6。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
21 Cover T,Hart P.最近邻模式分类。 IEEE信息论事务。 1967; 13:21–27. [ 谷歌学者 ] -
22 Chou KC,Wu ZC,Xiao X.iLoc-Euk:预测单复合体和复合真核蛋白亚细胞定位的多标记分类器。 《公共科学图书馆·综合》。 2011; 6:e18258。 doi:10.1371/journal.pone.0018258。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ] -
23 Keller JM、Gray MR、Givens JAJ。 一种快速的K-最近邻算法。 IEEE系统、人和控制论事务。 1985; 15:580–585. [ 谷歌学者 ] -
24 关于统计学中的广义距离。 印度国家科学研究院。 1936; 2:49–55. [ 谷歌学者 ] -
25 周朝中,张春涛。综述:蛋白质结构类的预测。 生物化学和分子生物学评论。 1995年; 30:275–349. doi:10.3109/10409239509083488。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
26 Liu T,Jia C.一种使用预测的二级结构信息的高精度蛋白质结构类预测算法。 理论生物学杂志。 2010; 267:272–275. doi:10.1016/j.jtbi.2010.09.007。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
27 Masso M,Vaisman II。 基于知识的计算突变用于预测人类非同义单核苷酸多态性的潜在疾病。 理论生物学杂志。 2010; 266:560–568. doi:10.1016/j.jtbi.2010.07.026。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
28 王涛,夏涛,胡XM。 预测膜蛋白类型的几何保持投影算法。 理论生物学杂志。 2010; 262:208–213。 doi:10.1016/j.jtbi.2009.09.027。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
29 Joshi RR,Sekharan S.蛋白质二级结构基序的特征肽。 蛋白质和肽字母。 2010; 17:1198–1206. doi:10.2174/0929686610792231500。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
30 Kandaswamy KK、Pugalenthi G、Moller S、Hartmann E、Kalies KU等。通过一种新的伪氨基酸组成模式,用遗传算法和支持向量机预测凋亡蛋白的位置。 蛋白质和肽字母。 2010; 17:1473–1479. doi:10.2174/0929866511009011473。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
31 Liu T,Zheng X,Wang C,Wang J.使用伪氨基酸成分预测凋亡蛋白的亚细胞位置:自协方差变换方法。 蛋白质和肽字母。 2010; 17:1263–1269. doi:10.2174/092986610792231528。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ] -
32 Mohabatkar H.使用Chou的伪氨基酸组成预测细胞周期蛋白。 蛋白质和肽字母。 2010; 17:1207–1214. doi:10.2174/092986610792231564。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]