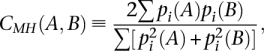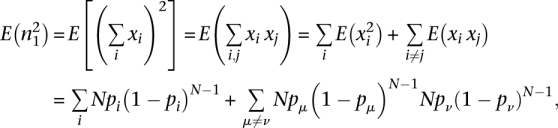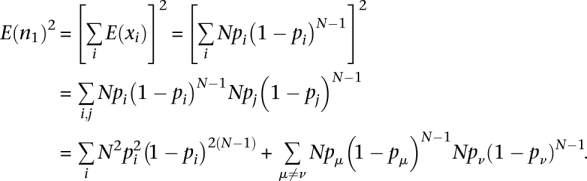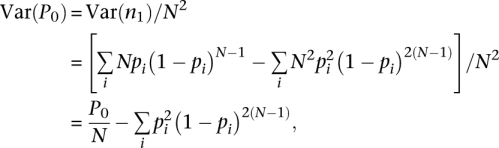摘要 我们提出了超高通量测序的强大应用,SAGE-Seq,用于准确定量正常和肿瘤乳腺上皮细胞转录组。 我们开发了数据分析管道,用于绘制线粒体和RefSeq基因的正反义链,库之间的标准化,以及识别差异表达的基因。 我们发现癌症转录组的多样性明显高于正常细胞。 我们的分析表明,转录物发现停滞在1000万读/样本,并建议所需的最小测序深度约为500万读。 SAGE-Seq与传统SAGE在正常和癌变乳腺组织上的比较显示,SAGE-Se对检测不太丰富的基因(包括编码已知乳腺癌相关转录因子和G蛋白偶联受体(GPCR)的基因)具有更高的敏感性。 SAGE-Seq能够识别传统SAGE无法识别的乳腺癌异常激活的基因和途径。 SAGE-Seq是鉴定人类疾病生物标志物和治疗靶点的有力方法。
微阵列和基于序列分析的技术已广泛用于基因表达谱分析,以创建细胞功能的全局图像( Adams等人,1991年 ; Schena等人,1995年 ; Velculescu等人,1995年 ). 早期的基因表达数据分析算法侧重于每种技术引入的偏见和局限性。 对于Affymetrix和NimbleGen微阵列等基于阵列的技术,已开发出克服探针特异性行为、GC含量偏差、染料偏差和交叉杂交的方法( Yang和Speed 2002 ; Johnson等人,2006年 ; Song等人,2007年 ). 而传统的基于序列分析的基因表达方法,如基因表达序列分析(SAGE)( Velculescu等人,2000年 ; 波利亚和里金斯2001 )和表达序列标签(EST)( Adams等人,1991年 )测序允许识别和量化已知基因和新基因,但测序吞吐量和成本严重限制了它们( Adams等人,1991年 ; Velculescu等人,1995年 ). 随着下一代测序平台以更低的成本提高吞吐量( Johnson等人,2007年 )将其应用于SAGE成为基因表达综合分析(SAGE-Seq)或其他应用的自然选择( Bloushtain-Qimron等人,2008年 )并保证更高的敏感性和特异性( Morrissy等人,2009年 ). 然而,SAGE-Seq在数据规范化、读取比对、差异表达基因的鉴定以及与传统SAGE的比较方面提出了独特的挑战。
为了解决上述问题,我们描述了数据分析管道,以处理在Illumina平台(以前称为Solexa)上对从正常和癌变人类乳腺组织样本中分离出的乳腺上皮细胞SAGE-Seq数据。 为了规范化不同库中的SAGE-Seq原始数据,我们使用非参数经验贝叶斯方法来减少序列采样偏差( 罗宾斯1956 ; 盖尔和桑普森1995 ). 评估数据集内和跨数据集的适当全球多样性度量,并将其用于对库进行聚类。 我们提出了一种映射策略,将SAGE-Seq标签与基因组对齐。 我们利用映射信息来最小化测序错误,并获得与RefSeq和线粒体基因相对应的正反义转录物的准确量化。 我们开发了一种鉴定具有统计学意义的差异表达基因的方法,并展示了其在正常和肿瘤乳腺上皮细胞差异基因检测中的实用性。 我们还比较了传统SAGE和SAGE-Seq数据集,并证明了SAGE-Seq在检测20倍以上差异表达基因方面的强大威力,具有更高的统计置信度。 路径分析表明,SAGE-Seq获得的更大测序深度允许识别出比传统SAGE多三倍多的具有统计学意义的基因本体论(GO)术语,并提高了它们的统计学显著性得分。 其中许多途径是SAGE-Seq新发现的,而传统SAGE完全忽略了这些途径。
结果 SAGE-Seq库生成 本研究中的SAGE-Seq文库由50000至100000个未培养的乳腺上皮细胞产生,这些细胞从正常健康女性的乳腺组织和原发性浸润性导管乳腺癌中分离( 表1 ). 细胞的免疫磁珠纯化和SAGE文库的生成基本上如前所述( Shipitsin等人,2007年 ),除非在Illumina平台上进行测序需要修改(参见方法)。 Illumina的原始数据由数百万个序列标签组成,但这里只有每次读取的前21个bp是有用的。 前4个bp都是“CATG”,这是SAGE文库构建过程中使用的NlaIII-mapping限制酶的识别位点。 MmeI被用作标记酶,将连接子中存在的识别位点的21 bp 3′立即剪切到NlaIII位点的5′。 因此,SAGE-Seq标签由5′“CATG”和17-bp独特的转录特异性序列组成。 跨面相关性表明SAGE-Seq库中丰度测量的高再现性(补充图S1)。
表1。
用于标签映射和排序错误最小化的管道 为了分析单个基因的表达,我们使用SeqMap( 江和黄2008 )并提出一个映射管道(补充图S3),将标签与RefSeq基因对齐。 这个映射管道允许我们将标签映射到RefSeq基因的线粒体、正反义转录本。 如果一个转录本有多个CATG,那么最靠近poly(a)尾部(3′端)的一个被称为最佳标记( 图1A ). 如果一个标记映射到多个RefSeq位置,并且只有一个标记是最佳标记,则最佳标记被视为唯一映射的位置。 否则,该标记是一个非唯一标记,其计数在映射的位置之间平均分配。 意义标签被定义为映射到已知转录物外显子意义链的标签。 反义标签是指不能映射为义标签,但能够映射到已知转录基因的反义链的标签( He等人,2008年 ).
图1。
SAGE-Seq标签对齐和排序错误最小化。 ( 一个 )最佳标记是指紧邻poly(A)尾部的最3′端NlaIII位点(CATG)的标记。 ( B类 )根据补充图S3中的标签对齐管道,样本N1的标签对齐统计。 其他数据集的详细映射如补充电子表格1所示。 ( C类 )标签映射期间显示的假定排序错误。 所有列出的标签都是唯一映射到同一RefSeq基因的最佳标签“ NM_001010号 。”(并不是在同一位置唯一映射到该基因的每个标签都被列出。) X(X) -axis是按标签计数的降序列出的标签序列。 最有可能是由于测序错误导致的序列中的一个碱基差异用红色标记。这个特定例子的测序错误最小化步骤是通过以下方式完成的:将所有这些标签的计数相加,并将其分配给标签“CATGGCCGTGTCCGCCTGCTA”,并删除所有其他标签。
映射结果还可用于识别排序错误,如所示 图1C 。我们将唯一映射到相同位置相同基因的标签计数合并,以减少因测序错误(测序错误最小化)而产生的噪音和采样偏差,从而减少后续差异表达基因分析中的假阳性数量。 参考基因组中的标签被用作测序误差最小化的一致标签。 例如,假设标签“GCCGTGTCCGCCTGCTA”出现190793次,它正好映射到参考基因组。 如果有3198个标签通过单个碱基对与此标签不同,在最小化测序错误后组合在一起,则总数为193961个; 因此,单碱基对失配的比例为1.6%(3198/193961)。 这相当于每基0.1%的测序错误率(17×0.001×[1−0.001] 16 = 1.7%). 这与Illumina高质量读数的估计一致( Shendure和Ji 2008 ). 以库N1为例,我们证明了大约76%的标签可以使用我们的管道进行唯一映射; 其中6%为线粒体标签,46%为唯一RefSeq感观最佳标签,14%为唯一感观非最佳标签,10%为唯一反义标签( 图1B ; 其他库的映射结果的补充电子表格1)。 所有后续分析都是在46%的独特感觉最佳标签上进行的。
正常和癌症转录组概述 细胞种群的基因表达模式在许多方面类似于生态系统中不同物种的种群,其中一个物种的个体就像我们研究中的转录本。 在典型的生态系统中,有些物种很丰富,而大多数物种很稀少( Magurran 2003年 ). 同样,SAGE-Seq分析数据显示,大多数基因表达水平较低(罕见转录物),少数基因表达量较大(大量转录物)( 图2A、B ). 有趣的是,尽管很少表达的标签占大多数,但在考虑其种群(表达水平)时,高度表达的独特标签仍占主导地位。 通过绘制标签计数占总标签计数的累计比例作为唯一标签计数的函数,我们发现,尽管只有一个计数的唯一标签占总标签数的63% S公司 (唯一标签的总数),它们只占 N个 (标签总数)( 图2B ). 问题是,这些低计数标签是由测序错误控制的假标签,还是以极低水平表达的真标签。 如上所述,在将序列错误最小化后,可以根据映射信息识别和纠正由于序列错误导致的一个不匹配的标签。 两个以上不匹配的标签仅占所有读取的0.01%(请参阅方法)。 因此,这些低计数标签不能用测序错误来解释,因为它们比这些错误所能解释的要丰富得多。 它们可能是低丰度转录物和核酸污染(可能是基因组DNA)的混合物,因为Sage-Seq制备协议不包括从RNA样品中消除基因组DNA的步骤。
图2。
独特标签计数频率图和非参数经验贝叶斯方法。 ( 一个 )库N1(黑色)和N5(红色)中唯一标记计数的频率。 X(X) -axis是观察到的标签计数 年 -axis是显示具有特定计数的唯一标记数的频率。 ( B类 )描述库N1中唯一标签分布的饼图:62.6%的唯一标签的标签计数1、12.1%、计数2、5.5%、计数3和19.8%的计数大于3。 外部图显示了唯一标记计数的累计分数。 尽管62.6%的唯一标签计数为1,但它们仅占总标签计数的3%。 ( C类 )标签比例散点图。 X(X) -axis是通过随机抽样10%的文库N1获得的伪文库1中的标签的比例。 Y(Y) -轴是通过随机抽样1%库N1获得的伪库2的平均比例。 数据点通过以下方式获得。 例如,在伪库1中查找比例为1×10的所有标签 −6 ,然后计算这些标记在伪库2中的平均比例,例如给出1×10 −5 。这给出了(1×10)处的数据点 −6 , 1 × 10 −5 ). 虚线为 年 = x 黑色符号表示使用最大似然估计值的比例,其中低表达和中等表达的标签(<100/百万)被高估,高表达的标签被低估(>1000/百万)。 红色符号表示使用非参数经验贝叶斯方法计算的比例,该方法在低丰度和高丰度标签中具有不同排序深度的两个库之间具有改进的、更具可比性的校正比例。
非参数经验贝叶斯归一化 如果每个SAGE库的测序深度相同(即相同 N个 ),不同库中的标记数可以直接比较。 然而,尽管大多数样品都进行了一条Illumina序列测定, N个 不同图书馆的数量从100万到1300万不等( 表1 ). 因此,为了准确比较不同文库的基因表达模式,需要对标签计数进行规范化。 一种直观的标准化方法是使用比例 第页 ,定义为 无 ,其中 n个 是唯一标记的计数。 被称为人口频率的最大似然(ML)估计量( 费希尔1922 ). 这种方法的缺点是 第页 测序数据中缺失的任何标记(未检测到的标记)被赋值为零,而它高估了 第页 低丰度和中丰度标记的数量,并低估了高丰度标记( 图2C ,黑色符号)。 此外,Illumina基因组分析仪的高通量高质量读数可以很好地估计不同富集水平下标签的群体频率( 图2A ). 作为先验应用的标签频率异质群体表明,对标签富集的最佳估计应该小于观察到的数量,因为低丰度转录本的群体大于丰度转录物的群体( 图2A ). 因此,需要一种更复杂的SAGE-Seq数据规范化方法。
我们应用了非参数经验贝叶斯(NEB)方法( 好1953 ; 罗宾斯1956 ; Orlitsky等人,2003年 )规范化具有不同排序深度的库(请参见方法)。 与ML相比,NEB有两个优势。首先,ML只是将未检测到的标签视为零,而NEB将未检测到的标签的比例估计为 P(P) 0 = n个 1 / N个 ,其中 n个 1 是计数为1的唯一标记的频率。 为了验证 P(P) 0 ,我们以1%的步长从1%到10%随机采样库N1,以生成10个具有不同测序深度的伪库。 我们使用NEB估算 P(P) 0 在每个伪文库中未检测到的标签中,将它们与原始文库中各自的比例进行比较,发现它们非常一致(补充图S2)。 其次,NEB根据观察到的计数和唯一标签计数频率分布的性质来调整标签计数( 图2A ),作为减少序列抽样偏差的经验先验( 盖尔和桑普森1995 ). 它还通过检测到的标签的估计总比例将调整后的比例重新规范化为1– P(P) 0 (有关NEB和ML标准化之间的比较,请参阅补充材料部分“差异表达基因的算法比较”)。 为了显示采样偏差的影响,我们从库N1中随机抽取10%(伪库1)和1%(伪库2)标签,生成两个序列深度相差10倍的伪库。 当比较两个伪库中标签的比例时,我们发现NEB标准化后的比例更具可比性,而ML高估了这一比例 第页 用于低计数标签和低估 第页 用于测序深度较低的伪库2中的高计数标记( 图2C ).
正常和癌症转录组的多样性 基于序列的基因表达谱的一个优点是它可以同时测量许多基因的绝对表达水平。 因此,我们可以获得细胞内以及库之间转录多样性的全局视图。 我们使用了两种不同的方法来比较我们分析的图书馆中的转录本多样性。 首先,我们使用了辛普森多样性指数(SID)( 辛普森1949 )描述每个文库中转录组的多样性。 SID捕获标记计数分布的方差,并且与排序深度无关(请参阅方法)。 较高的值表示较高的多样性,这意味着标签数量在不同基因之间的分布更广。 我们发现,在我们的数据集中,一般来说,癌症样本库的多样性高于正常样本库( 图3A、B ; Wilcoxon秩和检验, P(P) = 0.07284; 这个 P(P) -由于样本数量有限,值处于显著性边界)。 这种趋势可能是因为肿瘤表达更多的基因,要么是因为它们由更多不同的细胞群组成,要么是由于它们失去了维持组织和细胞类型特异性基因表达模式的正常表观遗传控制( 图4D ).
图3。
正常和癌症转录组的多样性。 ( 一个 )辛普森多样性指数用于衡量文库内基因表达多样性。 与正常组相比,癌症组的库内多样性较高。 ( B类 )方框图描绘了正常和癌症样本的辛普森多样性指数。 P(P) =0.07284(威尔科森秩和检验)。 ( C类 )定义为“1–Morisita-Horn相似性指数”的距离用于测量跨库的基因表达多样性。 正常组的图书馆彼此更相似,而癌症图书馆则更为多样化。 ( D类 )使用中定义的“距离”进行分层聚类 C类 分离正常和癌症库。
图4。
SAGE-Seq标记映射和测序深度饱和度曲线。 ( A–C )三个选定基因家族表达谱的差异覆盖率:转录因子( 一个 ),GPCR( B类 )和ABC运输车( C类 ). Y(Y) -axis列出了基因和 x -axis是平均基因表达指数(标准化标签计数的对数)。 红色和蓝色分别表示传统SAGE和SAGE-Seq。 SAGE-Seq在这些基因家族中检测到的基因比传统SAGE更多。 ( D类 )唯一的最佳标记基因数( 年 -轴)与测序深度相关( x -轴)。 最佳标记基因的数量是由最佳标记映射的唯一基因的数量,如果多个标记映射到同一基因的最佳标记,则计算为一个。 黑色和红色分别表示正常组和癌症组。 符号“○”和“▴”分别表示传统SAGE和SAGE-Seq。 实体曲线(饱和度曲线)是通过对正常(或癌症)组中所有库的组合进行采样而得到的模拟结果,这些库描述了随着测序深度增加的趋势。 传统SAGE识别的最佳标签基因比SAGE-Seq要少得多。 SAGE-Seq显示,癌症样本(红色三角形)比正常样本(黑色三角形)具有更多独特的最佳标记基因。 传统SAGE无法检测到这种差异(红圈与黑圈)。
第二种多样性是跨文库测量的,以研究不同个体的文库之间的基因表达多样性。 为了询问A和B两个库的相似程度,我们使用了Morisita-Horn(MH)相似性指数 C类 金属氢 ( 一个 , B类 ) ( 沃尔达1983 )(参见方法),并将其距离计算为 D类 ≡ 1 − C类 金属氢 ( 一个 , B类 ). 莫里西塔-霍恩指数比其他距离测量方法有几个优点。 首先,MH指数受以下因素影响不大 N个 和 S公司 ,这对于确定测量的差异不是由于测序深度的差异至关重要。 其次,与基于皮尔逊互相关的距离相比,对于标准差接近零的数据,MH指数没有奇异性。
我们发现,癌症样本不仅在每个个体中差异更大(辛普森指数),而且在不同个体中差异也更大(MH指数)( 图3C ). 这并不完全令人惊讶,因为正常细胞的生理作用在不同个体中基本相同,而肿瘤基因多样,在体内没有功能作用; 因此,没有选择压力来将其表型保持在一定范围内。 基于MH索引的库的层次聚类表明,癌症库之间的差异更大(库之间的距离更大),并且它们与正常库也有非常明显的区别( 图3D ).
SAGE-Seq与传统SAGE的数据质量比较 在读取比对和测序错误最小化之后,我们进一步评估了SAGE-Seq分析全基因组基因表达的能力,并将其与传统SAGE进行了比较。 随着测序覆盖范围的加深,SAGE-Seq在同一组内不同文库之间的数据相关性更高(补充图S4)。 此外,传统SAGE只能检测比例为10的基因 −5 到10 −3 ,而SAGE-Seq显示出更大的动态范围(定义为基因富集的检测范围),涵盖了从10到10的大约5个数量级 −7 到10 −2 例如,编码转录因子的基因通常以中或低水平表达,SAGE-Seq在我们的样本中检测到大约1300个转录因子的表达(在人类基因组中总共1658个转录因子中)。 传统SAGE检测到的大多数转录因子也通过SAGE-Seq检测到,而384个转录因子仅通过SAGE-Seq检测到( 图4A ). 我们观察到GPCR和ABC转运体基因编码的类似现象( 图4B、C ),已知其在正常细胞和癌细胞之间差异表达,并且以相对较低的水平表达( Li等人,2005年 ; 2009年院长 ).
为了确定已测序的SAGE-Seq库距离饱和的距离,我们计算了检测到的与每个库的测序深度相关的唯一最佳标记基因(唯一映射的最佳标记)的数量。 唯一映射的最佳标签的数量是检测到的基因数量的一个良好指标。 深度测序有望检测到更多的基因,直到检测到所有基因时达到一个平台。 为了克服缺乏涵盖广泛测序深度的数据,我们合并了所有癌症(或正常)文库,并分析计算了在不同测序深度检测到的唯一标签基因的数量,以获得饱和度曲线( 图4D ; 见分析计算方法)。 对于测序深度低于300万读,检测到的基因数量随着测序深度(快速增长区域)急剧增加。 该速率继续以低于500万次读取的较慢速度(缓慢增长区域)增长,直到正常和癌症样本的读取量都达到1000万次左右为止( 图4D ). 这表明SAGE-Seq的理想测序深度应在1000万读以上,每个库的最小期望序列深度为500万。 Sage-Seq数据点(三角形)都靠近或位于生长缓慢的区域,在那里大多数转录组都被测序。 传统的SAGE数据点(圆圈)仍处于快速增长区域,由于测序深度较低,超过一半的转录组未被检测到。 图4D 还表明癌症(红三角)样本中表达的基因比正常(黑三角)样本多,这与我们之前的发现一致,即癌症样本在每个库内和库间具有更高的转录多样性。
采样噪声和生物变异性 为了检测两种条件下差异表达的基因,了解基因表达变异的来源很重要。 SAGE-Seq或任何使用测序技术的技术的变异性的主要来源是取样变异; 大多数分析传统SAGE数据的算法使用各种方法解决了这一问题( Velculescu等人,1995年 ; Cai等人,2004年 ). 基于序列的转录组分析可以建模为二项式采样过程,替换近似泊松分布,因为使用当前技术( Kharchenko等人,2008年 ),测序转录物是测序器上加载的cDNA总量的极小一部分。 如果同一个文库被多次测序,泊松模型表明特定基因标签数的方差等于其丰度。
当检查正常文库中基因的经验方差与其各自的归一化计数(由NEB归一化并缩放到相同的测序深度 N个 =100万),观察到的差异确实取决于其基因表达水平(计数)( 图5A ; 带斜率的红色虚线 α 正面 ≈2.0(对数图中)。 然而,如果我们将随机二项抽样的方差-平均斜率表示为α 苏格兰皇家银行 ,根据泊松分布预计为1.0( 苏格兰皇家银行 代表 第页 安多姆 b条 诺米亚的 秒 采样,中的蓝色虚线 图5A ),我们观察到过度分散( α 正面 ≈2α 苏格兰皇家银行 > α 苏格兰皇家银行 =1),这意味着观测数据的过度可变性明显大于随机参考模型(本例中为泊松模型)中的预期可变性。 这表明基因表达变异性对平均表达水平具有非线性依赖性,这表明在我们的数据集中,过度分散可能是除采样变异外,生物个体变异的结果(见方法)。 我们还观察到正常组和癌症组中管家基因子集和唯一映射的最佳标记子集之间的过度分散(数据未显示)。
图5。
差异表达基因及其变异。 ( 一个 )去除噪声和归一化后,七个标准库的均值-方差图。 红色虚线是对数图中最佳的线性拟合。 斜率给出了指数 α 正面 ≈1.9蓝色虚线是通过采样引入的均值-方差线。 ( B类 )鉴别差异表达基因的流水线:(1)测序误差最小化:标签对齐后,在相同位置映射到相同基因的标签被组合在一起; (2) NEB用于规范不同序列深度的不同库; (3) 过滤以删除少于两个库中计数≥3/百万的标记,然后是日志 2 转型; (4) SAM用于检测差异表达基因。 ( C类 )检测到差异表达基因( 顶部 )和激活的途径( 底部 )在SAGE-Seq和传统SAGE中。 SAGE-Seq以1%的FDR识别约4000个差异基因,而传统SAGE以更宽松的截止值(10%的FDR)识别<200个差异基因。 在 P(P) =0.001,SAGE-Seq确定了99条在乳腺癌中显著激活的通路,而传统SAGE仅显示32条。 仅由SAGE-Seq识别而被传统SAGE遗漏的80条通路均为乳腺癌相关通路。 ( D类 )重叠比率(定义为重叠基因数除以顶部传统SAGE中的基因数 x 差异表达基因的百分比,其中 x 在0和1之间更改。 黑色符号表示实际数据(SAGE-Seq与传统SAGE)。 这表明SAGE-Seq和传统SAGE之间的顶级差异表达基因列表几乎没有重叠。 红色符号表示模拟(SAGE-Seq与向下采样的SAGE-Seq)。 抽样SAGE-Seq意味着从每个SAGE-Seq库中对50 k个标签进行二项抽样; 50000是传统SAGE的典型测序深度。 模拟证实了与实际数据相同的结论:与传统的SAGE相比,SAGE-Seq给出了不同的差异表达基因列表。 更深入的测序表明,传统SAGE与SAGE-Seq相比识别出不同组的差异表达基因,从而证实了我们的结论,即传统SAGE缺乏足够的测序深度。
差异表达基因的分析 转录组分析的主要应用之一是识别不同样本之间差异表达的基因。 在标签对齐和测序错误最小化后,我们的分析管道用于识别差异表达基因( 图5B )首先应用非参数经验贝叶斯方法作为归一化步骤,以减少采样偏差,并使不同的库具有相同的测序深度( N个 =100万; 归一化测序深度对差异基因没有影响,这与文库的测序深度不同。)。 归一化后,所有库中计数≥3/百万in≤2的标签被丢弃。 这有效地删除了大量非信息性标签,这些标签要么包含离群值,要么计数太低,无法检测具有统计意义的差异表达式,并在后续分析中节省了计算时间和存储空间。
然后应用对数变换获得表达指数,并解耦观察到的方差和基因平均表达水平之间的相关性( 图5A ). 从数量上讲,我们库中观察到的方差与表达式级别的平方成正比。 根据统计学中的delta方法,对数变换是稳定方差的正确变换(参见方法)。 另一种转换是 电弧(电弧) ,这也是一种类似对数的变换,但具有零处无奇点的优点( Huber等人,2002年 ). 补充图S5显示,在对归一化计数应用以2为基数的对数转换后,对于中等丰度和高丰度标记,表达指数的方差几乎与其平均值无关。 最后,将SAM(微阵列显著性分析)算法应用于两组样本的表达指数,以识别差异表达的基因( 图5B ; Tusher等人,2001年 ). 我们也尝试了标准 t吨 -测试并发现许多误报,这些误报是由于低估了经验标准差而导致的极端情况 t吨 值。 SAM算法稳定方差以减少误报。 此步骤中可以使用其他统计测试,而不是使用SAM,如Robinson和Smyth的调节 t吨 -测试或Baggerly’s t吨 w个 测试( Baggerly等人,2003年 , 2004 ; Lu等人,2005年 ; Robinson和Smyth 2007 ). 分析差异表达基因的另一种选择是使用过度分散的模型,如过度分散的logistic回归或过度分散的对数线性模型( Baggerly等人,2004年 ; Lu等人,2005年 ). 然而,这些基于模型的方法是否可以扩大到SAGE-Seq数据的更深测序深度,需要通过更多数据的系统分析进行验证。
我们比较了SAGE-Seq和传统SAGE中正常和癌症的差异表达基因列表。 SAGE-Seq和传统SAGE分别检测10052和4953个最佳标记基因的表达(即存在)(补充电子表格2),99%(4904)重叠。 我们使用 问 -Storey和 Tibshirani(2003) 传统的SAGE序列不够深,不允许类似的 P(P) -值或FDR截止值,如SAGE-Seq。 SAGE-Seq在1%FDR下鉴定出约4000个差异表达的最佳标签基因,而传统SAGE在10%FDR下检测到的少于200个( 图5C ). 更深入的测序可以提高SAGE-Seq的统计能力,以检测更多差异表达的基因。 为了比较这两个差异表达基因列表,我们检查了基于它们的基因排序 t吨 -分数。 前10%的基因 t吨 -分数(传统SAGE的495个基因和SAGE-Seq的1005个基因)被用作差异表达基因列表,以比较这两种方法。 根据先前的研究,SAGE-Seq检测到所有26个已知在正常和乳腺癌样本中差异表达的基因,而传统SAGE仅检测到四个(补充表S1)。
令人惊讶的是,当比较两种方法识别出差异表达的前10%的基因之间的重叠时,我们只识别出54个基因。 进一步分析证实,传统SAGE和SAGE-Seq检测到的最高差异表达基因差异很大( 图5D ; 黑色符号)。 许多因素都可能导致这种差异,例如文库制备方案和样本的差异。 除这些因素外,我们还观察到,SAGE-Seq检测到的差异表达最高的基因通常以中等或低水平表达(~100/百万;参见补充图S7),传统SAGE要么完全无法检测到,要么标签计数太低(两个或三个),无法显示具有统计能力的差异表达。 SAGE-Seq中这些差异表达的标签不太可能来自基于观察到的标签计数的测序错误。 这些数据表明,SAGE-Seq测序深度的增加导致检测到一组不同的差异表达基因。 为了证明这一点,我们采用了模拟方法,因为使用从原始乳腺组织中分离的细胞数量有限的特定细胞群,不允许从同一样本中生成SAGE-seq和传统SAGE库。 我们选取了14个SAGE-Seq库,并对其进行了抽样(二项式抽样),以达到传统SAGE的序列深度水平(~50000)。 这些模拟文库的顶部差异表达基因也与原始SAGE-Seq文库几乎没有重叠( 图5D ,红色符号)。
正常和癌症样本之间激活的路径和网络不同 为了确定哪些信号通路被SAGE-Seq和传统SAGE识别为差异激活,我们使用MetaCore对差异表达基因集进行了基因本体和通路分析的组合( Nikolsky等人,2009年 ). 然而,SAGE-Seq在1%的FDR截止值下识别出3587个差异表达基因,而传统SAGE识别出的差异表达最显著的基因的FDR>9%。 因此,我们决定将传统SAGE基因(493)和SAGE-Seq基因识别出的前10%差异表达基因取为1%FDR(3587),因为在传统SAGE中,FDR截止值导致差异表达基因太少(补充电子表格3)。 MetaCore提供了 P(P) -每个测试GO术语或路径名称的值。 使用 P(P) -值为10 −3 SAGE-Seq确定了99条重要途径作为显著性的截止点,而传统SAGE只有32条重叠19条( 图5C ; 补充电子表格4)。 SAGE-Seq和传统SAGE之间通常富含以下途径和GO过程:凋亡、细胞粘附、细胞骨架重塑、发育、免疫反应、G蛋白信号传导、信号转导和转录。 这些都是已知的与乳腺癌相关的途径,SAGE-Seq在每个类别中都确定了具有较高统计意义的术语。 根据已发表的文献,SAGE-seq而非传统SAGE确定的80个额外的重要GO类别均与癌症相关,通常或特别与乳腺癌相关,尤其是诸如凋亡和存活、细胞周期、雄激素受体信号、TGFB信号、NFKB信号、, 巴西航空公司1 -介导的DNA损伤、p53信号通路、发育和细胞周期调控 ESR1系列 和 欧洲标准2 (雌激素受体)、G蛋白信号传导以及翻译和转运途径。 这些途径中的基因通常以低水平表达,这与 图4、B和C 显示了SAGE-Seq检测到的GPCR和ABC转运蛋白家族中的许多基因,但传统SAGE检测不到这些基因( Li等人2005 ; 2009年院长 ). 特别值得注意的是,NFKB和TGFB通路出现在多个GO和通路分支中,已知在乳腺癌中受到差异调节( Shipitsin等人,2007年 )发现在SAGE-Seq中显著,但在传统SAGE中不显著。
讨论 在本研究中,我们系统地评估了SAGE-Seq的转录组分析及其识别正常和肿瘤乳腺上皮细胞之间差异表达基因的能力。 我们是第一个应用NEB方法对不同的高通量SAGE-Seq库进行规范化,以纠正由于不完全采样导致的采样偏差。 NEB归一化可应用于基于随机抽样的其他类型技术,如RNA-seq。 我们设计了一条管道,将SAGE标签与正反义转录物对齐,通过标签对齐将测序错误降至最低,并提出了一种通过同时考虑采样和生物变异性来检测差异表达基因的方法。 我们比较了SAGE-Seq和传统SAGE,以检查测序深度对基因覆盖率和差异表达基因检测的影响。 对正常乳腺上皮细胞和肿瘤乳腺上皮细胞之间的SAGE-Seq数据进行比较后发现,乳腺癌与正常乳腺细胞相比,具有更高的细胞内和跨细胞多样性。 SAGE-Seq在比传统SAGE(10%FDR)严格10倍的临界值(1%FDR)下识别出20倍以上的差异表达基因,以及3倍以上的乳腺癌特异性激活途径,表明其更高的敏感性和特异性。
识别与生理过程相关的基因表达变化是生物学的中心问题,尤其是在人类疾病的研究中( Zhu等人,2008年 ). 常用的方法包括EST测序、cDNA微阵列杂交、减法克隆、差异显示和基因表达的序列分析(传统SAGE)( Adams等人,1991年 ; Schena等人,1995年 ; Velculescu等人,1995年 ). 与基于阵列的杂交方法相比,SAGE-Seq具有许多优点。 首先,SAGE-Seq具有更高的灵敏度,可以以高置信水平检测不太丰富的基因。 其次,SAGE-Seq不太容易受到诸如探针效应和杂交偏差等技术伪影的影响( Yang和Speed 2002 ). 第三,SAGE-Seq不需要分析转录本的先验知识; 因此,它可以对细胞中的转录组进行全局分析。
在微阵列数据中观察到高表达基因表达水平的过度分散,因此,分析通常在对数强度水平上进行( Irizarry等人,2003年 ). 然而,大多数人将过度分散归因于探针杂交和芯片平台固有的扫描偏差。 我们定量地确定了生物变异性和平均表达水平之间的关系。 这里提供的SAGE-Seq数据不仅表明许多SAGE分析算法中使用的泊松分布不足以捕获生物方差,而且还表明生物样本中丰富的基因具有较高的变异性( 图5A ). 这表明细胞能够更好地耐受高表达基因水平的变化。 这些发现还意味着,如果将重点放在中丰度或低丰度转录物上,疾病标记物和药物靶点的发现可能会更有成效,因为这些转录物显示同一组织类型内样本之间的差异较小,并且它们的表达差异可能在疾病过程中发挥更重要的作用。
SAGE-Seq具有更深的测序深度,能够检测到比传统SAGE更显著的差异表达转录物。 SAGE-Seq鉴定出的差异表达最高的基因不是最丰富的基因,而是在中或低水平(~100/百万)表达。 对于传统的SAGE,它的测序深度要小20倍,这些标签将处于被检测的边界。 因此,传统SAGE无法在不同条件下区分这些基因。 同时,这些不太丰富的基因通常是转录因子和受体,在细胞调节和肿瘤发生中发挥重要作用( 图4A–C ). 因此,即使这些基因表达的微小变化也可能对整个细胞环境产生显著影响。 似乎数量较少的基因变异性也较小( 图5A )这使得它们能够被检测为差异表达最高的基因,尽管它们表达水平的绝对变化不是最大的。 因此,高通量测序技术提供了一个机会,可以更详细地揭示基因表达的细微变化,并提高统计能力。
总之,我们在这里表明,SAGE-Seq是一种强大且具有成本效益的方法,可以对从原始人类组织样本中分离的少量细胞进行基因表达谱分析,我们还提供了数据分析工具,使研究人员能够解读大量SAGE-Seq数据集的生理意义。
方法 SAGE-Seq库建设 我们将SAGE-Seq库生成的详细协议发布在 http://research4.dfci.harvard.edu/polyaklab/protocols_linkpage.php .
本研究中的所有SAGE和SAGE-Seq文库均由刚从人类乳腺组织样本中分离的免疫磁珠纯化细胞生成; 因此,细胞数量是根据10微升体积内捕获细胞数量的显微镜检查估计的,它们在50000–100000个细胞范围内。 然而,根据FACS分析和相同细胞类型的分类,我们知道它们在组织样本中的大致丰度。 所有细胞都被直接裂解和处理以进行聚(A)RNA选择,然后进行文库制备。 聚(A)RNA的数量可以通过纳米滴或SYBR绿色II测量,但如果细胞数量非常有限,我们只需直接进行文库准备。 据估计,一个细胞含有10 pg的总RNA,100000个细胞含有~100 ng的总RNA和~1–10 ng的聚(A)RNA(取决于细胞类型,肿瘤细胞通常具有较高的RNA含量/细胞)。 此外,在继续SAGE-Seq样品制备之前,保存10%的poly(A)RNA用于细胞纯度的半定量RT-PCR测试,这也给出了对存在的可转录mRNA的估计。
排序错误最小化 在每基0.1%的错误率下,没有错误的标签数量为(1−0.1%) 17 =0.9831353,一个错误的标签总数为17×0.001×(1−0.001) 16 = 0.01673003. 因此,至少有两个碱基存在错误的标签总数为:1−0.9831353−0.01673003=0.0001346471。
辛普森多样性指数 辛普森多样性测量( 辛普森1949 )(SID)定义为:
哪里 n个 我 是的计数 我 第个标签和 N个 是标记计数的总数。 SID=1表示一个标签支配系统的所有标签计数,这意味着没有多样性(最高支配)。 SID值越大,多样性越高(优势度越低)。 仿真证实,SID不受测序深度的强烈影响。
Morisita-Horn相似指数 Morisita-Horn相似指数, C类 金属氢 ( 一个 , B类 ),在两个库之间, 一个 和 B类 ,定义为:
哪里 第页 我 ( 一个 )以及 第页 我 ( B类 )是基因(或标签)的比例 我 用于库 一个 和 B类 分别是。 MH相似性指数与测序深度无关 N个 .
抽样方差和生物方差 高通量测序技术被建模为具有替换的二项式采样过程。 混合不同样本的生物可变性,由于采样,引入了另一层可变性。 定义 第页 n个 、带计数的标签比例 n个 作为, 第页 = n个 / N个 ,其中 N个 是标记计数的总数。 从我们的层次模型来看,平均比例 基因丰度是基因真实丰度的无偏估计量。 然而, 秒 2 ( x 我 ),标度计数的观测方差 x 我 基因的 我 是个体真实生物方差和抽样方差的相加(分析证明的补充材料),
使用非参数经验贝叶斯校正的归一化 我们使用简单的Good-Turing估计器(SGT)实现经验贝叶斯( 盖尔和桑普森1995 ). 假设mRNA中所有唯一标签的总数为 秒 、和 第页 我 是标签的真实比例 我 ,这是我们想要从数据中估计的。 观测标签数的经验Bayes估计 第页 是:
哪里 n个 第页 是带有count的标记数 第页 。因此,每个代表的所有标记的预期总机会 第页 次( 第页 ≥1)为:( 第页 + 1) n个 第页 + 1/ N个 ,其中 N个 是测序深度( N个 = n个 1 + 2 n个 2 + 3 n个 三 +…). 因此,样本中所有标签的预期总概率为:(2 n个 2 + 3 n个 三 +…)/ N个 = 1 − n个 1 / N个 在SGT中,未检测到的标签的比例, P(P) 0 估计为
哪里 n个 1 表示计数为1的唯一标记的数量(频率)。 SGT后修正的标签总数, N个 *,是 N个 * = Σ n个 第页 第页* .基因与计数的比例的经验贝叶斯估计 第页 , 第页 第页 *,被重新规范化 N个 *作为
不可见标签的良品期估计方差 的方差 P(P) 0 可以用以下公式计算:Var( P(P) 0 )=变量( n个 1 )/ N个 2 . n个 1 = Σ 我 无 我 (1- 第页 我 ) N个 − 1 在二项抽样近似的假设下。 引入新的随机数 x 我 : x 我 =1,如果 我 第个标签在排序深度仅使用一个标签进行排序 N个 和 x 我 = 0 否则。 然后:
和
因此,
它给出了:
这表明未发现标签的Good-Turing估计器是一个稳定的估计器,如补充图S2所示。
饱和度曲线计算 我们将mRNA中所有独特标签的总数定义为 秒 和 第页 我 作为标签的真实比例 我 . 第页 我 是根据数据估计的。 因此,根据二项式抽样,未发现标签的平均值为:
其中,总和是可能的唯一最佳标记和 N个 是测序深度。 因此,检测到的唯一最佳标记基因的数量是唯一最佳标记的基因总数减去 n个 0 ( N个 )如所示 图4D .
方差稳定化 对于我们数据中观察到的方差-均值关系,稳定方差的正确转换是基于delta方法的对数转换。 我们提供以下证据。 假设一个随机变量 x 遵循平均值分布 μ 和方差 σ 2 .考虑转换 克 ( x ). 泰勒展开式 克 ( x )周围 μ 第一个订单是 克 ( x )≈ 克 ( μ ) + ( x − μ ) 克 ′( μ ). 因此,转换后的变量 克 ( x )具有近似平均值 克 ( μ )和近似方差Var[ 克 ( x )] ≈ σ 2 [ 克 ′( μ )] 2 。在我们的数据中, μ 和 σ 2 满足观察到的依赖性 σ 2 ∼ μ 2 ,产生Var[ 克 ( x )] ≈ μ 2 [ 克 ′( μ )] 2 .假设转型 克 稳定方差,Var[ 克 ( x )]是独立于 μ ,因此 克 ′( μ ) = c(c) / μ ,其中 c(c) 是一个常数。 在以下方面进行整合 μ 给出了稳定化变换的形式 克 应该是: 克 ( x )=对数 x .
致谢 我们感谢Andrea Richardson(Brigham and Women's Hospital)在获取乳腺肿瘤样本方面的帮助; 黄海燕(Haiyan Huang)、李才(Li Cai)、王莫林(Molin Wang)和戴维·哈林顿(David Harrington)进行了有价值的讨论; 喜欢尼克森的英语校对。 这项工作得到了Dana-Farber女性癌症项目之友(X.s.L.)、NIH R01 1HG004069(X.s.L.)、NCI P50 CA89393 CA和R01 CA116235-04S1(K.P.)、AVON基金会(K.P.)、Terri Brodeur乳腺癌研究基金会(s.C.)和Susan G.Komen基金会PDF0707996(M.s.,R.M.)的支持。
作者贡献: Z.W.进行了计算分析。 Z.W.、X.S.L.和K.P.设计了这项研究并撰写了手稿。 Z.W.、X.S.L.、C.M.、A.S.和J.L.开发了分析方法。 S.C.、M.S.、R.M.、M.B.和T.N.进行了实验并分析了数据。 S.S.为研究提供了正常组织样本。
工具书类
Adams MD、Kelley JM、Gocayne JD、Dubnick M、Polymeropoulos MH、Xiao H、Merril CR、Wu A、Olde B、Moreno RF等,1991年。 互补DNA测序:表达序列标签和人类基因组计划。 科学252:1651–1656 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Baggerly KA、Deng L、Morris JS、Aldaz CM 2003。 SAGE中的差异表达:解释库变异之间的正常现象。 生物信息学19:1477–1483 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Baggerly KA、Deng L、Morris JS、Aldaz CM 2004。 SAGE的过度分散逻辑回归:建模多组和协变量。 BMC生物信息学5:144 doi:10.1186/1471-2105-5-144 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Bloushtain-Qimron N、Yao J、Snyder EL、Shipitsin M、Campbell LL、Mani SA、Hu M、Chen H、Ustyansky V、Antosiewicz JE等,2008年。 人类乳腺中细胞类型特异性DNA甲基化模式。 《国家科学院院刊》105:14076–14081 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Cai L、Huang H、Blackshaw S、Liu JS、Cepko C、Wong WH 2004。 使用泊松方法对SAGE数据进行聚类分析。 基因组生物学5:R51 doi:10.1186/gb-2004-5-7-51 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Dean M 2009年。 ABC转运体、耐药性和肿瘤干细胞。 乳腺生物肿瘤杂志14:3-9 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
费希尔RA 1922。 理论统计学的数学基础。 菲洛斯Trans R Soc Lond 222:309–368 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Gale WA,Sampson G,1995年。 良好的频率估计,无撕裂。 凌志军2:217–237 [ 谷歌学者 ]
1953年国际司法日。 物种的种群频率和种群参数的估计。 生物医学40:237–264 [ 谷歌学者 ]
He Y、Vogelstein B、Velculescu VE、Papadopoulos N、Kinzler KW 2008。 人类细胞的反义转录体。 科学322:1855–1857 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Huber W、von Heydebreck A、Sültmann H、Poustka A、Vingron M,2002年。 方差稳定应用于微阵列数据校准和差异表达的量化。 生物信息学18:S96–S104 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Irizarry RA、Hobbs B、Collin F、Beazer-Barclay YD、Antonellis KJ、Scherf U、Speed TP 2003。 高密度寡核苷酸阵列探针水平数据的探索、规范化和总结。 生物统计学4:249–264 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
姜浩,王伟,2008。 SeqMap:将大量寡核苷酸映射到基因组。 生物信息学24:2395–2396 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Johnson WE、Li W、Meyer CA、Gottardo R、Carroll JS、Brown M、Liu XS,2006年。 ChIP-ChIP平铺阵列的基于模型的分析。 国家科学院院刊103:12457–12462 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Johnson DS、Mortazavi A、Myers RM、Wold B,2007年。 体内蛋白质-DNA相互作用的全基因组绘图。 科学316:1497–1502 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Kharchenko PV,Tolstorukov MY,Park PJ 2008。 DNA结合蛋白ChIP-seq实验的设计和分析。 国家生物技术26:1351–1359 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Li S,Huang S,Peng SB 2005年。 癌细胞中G蛋白偶联受体的过度表达:参与肿瘤进展。 国际癌症杂志27:1329–1339 [ 公共医学 ] [ 谷歌学者 ]
Lu J,Tomfohr JK,开普勒TB 2005。 识别多个SAGE库中的差异表达:一种过度分散的对数线性模型方法。 BMC生物信息学6:165 doi:10.1186/1471-2105-6-165 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Magurran AE 2003年。 物种的共性和稀有性。 《测量生物多样性》,第18页,新泽西州霍博肯市威利-布拉克韦尔 [ 谷歌学者 ]
Morrissy AS、Morin RD、Delaney A、Zeng T、McDonald H、Jones S、Zhao Y、Hirst M、Marra MA,2009年。 用于癌症基因表达谱分析的下一代标签测序。 基因组研究19:1825–1835 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Nikolsky Y、Sviridov E、Yao J、Dosymbekov D、Ustyansky V、Kaznachev V、Dezso Z、Mulvey L、Macconaill LE、Winckler W等,2008年。 人类乳腺癌中扩增和突变基因之间的全基因组功能协同作用。 癌症研究68:9532–9540 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Nikolsky Y、Kirillov E、Zuev R、Rakhmatulin E、Nikolskaya T,2009年。 在一个集成的“基于知识的”平台中对OMIC数据和小分子化合物进行功能分析。 方法分子生物学563:177-196 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Orlitsky A,Santhanam NP,Zhang J 2003。 永远好的图灵:渐近最优概率估计。 科学302:427–431 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Polyak K,Riggins GJ 2001。 利用基因表达技术的系列分析进行基因发现:对癌症研究的意义。 临床肿瘤学杂志19:2948–2958 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Robbins H 1956年。 统计的经验贝叶斯方法。 第三届伯克利数理统计与概率研讨会论文集,第1卷,第157-163页,加利福尼亚大学出版社,加州伯克利 [ 谷歌学者 ]
Robinson医学博士,Smyth GK,2007年。 用于评估标记丰度差异的适度统计测试。 生物信息学23:2881–2887 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Schena M、Shalon D、Davis RW、Brown PO 1995。 用互补DNA微阵列定量监测基因表达模式。 科学270:467–470 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Shendure J,Ji H 2008。 下一代DNA测序。 国家生物技术26:1135–1145 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Shipitsin M、Campbell LL、Argani P、Weremowicz S、Bloushtain-Qimron N、Yao J、Nikolskaya T、Serebryskaya T、Beroukhim R、Hu M等,2007年。 乳腺肿瘤异质性的分子定义。 癌细胞11:259–273 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
辛普森EH 1949。 多样性测量。 性质163:688 [ 谷歌学者 ]
Song JS、Johnson WE、Zhu X、Zhang X、Li W、Manrai AK、Liu JS、Chen R、Liu XS,2007年。 基于模型的双色阵列分析(MA2C)。 基因组生物学8:R178 doi:10.1186/gb-2007-8-8-R178 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Storey JD Tibshirani R 2003。 全基因组研究的统计意义。 美国国家科学院院刊100:9440–9445 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Tusher VG,Tibshirani R,Chu G,2001年。 微阵列用于电离辐射反应的显著性分析。 国家科学院院刊98:5116–5121 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Velculescu VE、Zhang L、Vogelstein B、Kinzler KW 1995。 基因表达的系列分析。 科学270:484–487 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Velculescu VE、Vogelstein B、Kinzler KW 2000。 用SAGE分析未知转录组。 趋势Genet 16:423–425 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
沃尔达H 1983。 多样性、多样性指数和热带蟑螂。 《生态》58:290–298 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
杨玉华,速度T 2002。 cDNA微阵列实验的设计问题。 Nat Rev Genet 3:579–588 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Zhu J,Zhang B,Schadt EE 2008。 药物发现的系统生物学方法。 先进基因60:603–635 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Genome Research的文章由以下机构提供 冷泉港实验室出版社