摘要 背景 淀粉样β前体蛋白(APP)转基因小鼠和阿尔茨海默病(AD)患者的主动和被动免疫治疗均显著减少了淀粉样斑块的累积,尽管淀粉样斑块退化的程度存在很大差异。 对9名临床诊断为AD痴呆的患者主动免疫aβ肽1-42(AN-1792),并进行详细的死后生化分析。 将这些患者与6例非免疫性AD病例和5例非痴呆对照(NDC)病例进行比较。
结果 对所有患者进行AD病理学评估,包括淀粉样斑块、神经纤维缠结和血管淀粉样变性。 这项研究表明,两名免疫治疗受者因AD以外的其他疾病而患有痴呆症。直接神经病理学检查始终显示淀粉样斑块明显受损的小到广泛区域。 通过ELISA对Aβ物种残留物的特征分析表明,总的Aβ水平可能已经降低,尽管由于治疗个体中Aβ肽的数量变化很大,这些数据必须视为暂时性的。 色谱分析和蛋白质印迹显示了丰富的二聚体Aβ肽。 SELDI-TOF质谱显示了大量aβ相关肽,其中一些具有较长的C末端序列。 与NDC和非免疫AD组相比,免疫AD患者灰质中的促炎性TNF-α水平显著增加。
结论 免疫治疗反应具有极强的可变性。 考虑到表征衰老的广泛生物变异,并使可靠AD生物标记物的识别复杂化,这种差异将使流行病学和治疗研究结果的解释具有挑战性。 尽管在某些情况下,AN-1792明显去除了淀粉样斑块,但AD临床进展的比例变化并不明显。 斑块消除并没有改变痴呆症的下降轨迹,这表明这些沉积物本身可能不是痴呆症的根本原因。
背景 阿尔茨海默病(AD)痴呆症影响着全球2600多万老年人,预计到2050年,这一数字将翻两番[ 1 ]. 仅在美国,就有530万人患有AD,估计每年花费1720亿美元[ 2 ]. 鉴于这些令人震惊的流行病学数据,迫切需要制定战略和治疗干预措施,以预防、减轻或延迟这种痴呆症的发病年龄。
自Glenner和Wong的开创性论文发表以来,AD患者大脑中淀粉样β(Aβ)肽的沉积一直是密集研究的主要焦点[ 三 ]和Masters等人[ 4 ]. 观察AD脑实质和血管Aβ肽的大量积累被纳入淀粉样级联假说,作为AD痴呆发病的中心致病因素[ 5 , 6 ]. 淀粉样β前体蛋白(APP)、早老蛋白(PS)1和PS2突变的遗传和生物化学研究均增强淀粉样蛋白沉积,有力地支持了这一假设。 对于许多AD研究者来说,淀粉样蛋白假说已经达到了虚拟教条的地位。 然而,也有持不同政见者对这一强有力的原则提出了批判性的质疑[ 7 , 8 ].
APP和Aβ肽是进化上保守的多功能分子。 有人认为Aβ可能在神经干细胞的发育中发挥神经原功能[ 9 ]. 还假设Aβ与神经毒性物质结合,淀粉样变刺激其吞噬细胞清除,从而代表对损伤的生理反应[ 10 , 11 ]. β-和γ-分泌酶活性下降会减少Aβ的生成,从而导致神经元死亡[ 12 ]. Aβ肽是小胶质细胞活化的强大调节剂[ 13 , 14 ],具有血管收缩活性[ 15 ]可能在神经炎症和抑制血管生成中起保护作用[ 16 ]. 沉积在脑微血管壁上的Aβ也有止血功能[ 17 , 18 ].
转基因(Tg)小鼠模型已被设计用于表达特征良好的APP、PS和tau突变。 这些小鼠被广泛用于测试各种旨在减轻Aβ肽积累的有害影响或促进其特定清除的化合物和策略的功效。 特别关注淀粉样肽免疫疗法,以确定其对病理和认知的影响。 Tg小鼠的主动和被动免疫治疗均成功减少了淀粉样斑块积聚和认知缺陷(参考文献综述[ 19 ]). 不幸的是,这些有希望的干预措施尚未在人体临床试验中产生明确的疗效。 一些因素可能解释了Tg动物和人类AD患者对淀粉样蛋白免疫治疗的反应之间的差异。 转录组分析表明,小鼠对衰老的反应与人类截然不同[ 20 ]. 此外,寿命、固有衰老率、小鼠大脑相对简单以及人工施加在小鼠模型上的诱导损伤性质的差异也可能是这些差异的原因[ 21 - 23 ].
痴呆患者的多项临床和病理观察,包括淀粉样斑块的生化成分,表明AD的发病机制是一个复杂的多因素过程,涉及aβ以上。 支持这些原则的是对Aβ免疫个体的大脑进行的神经病理学、生物化学和免疫化学研究。 虽然有明显的大范围区域没有淀粉样斑块,但血管淀粉样蛋白和可溶性Aβ的数量明显增加,而神经原纤维缠结(NFT)不受影响[ 24 - 31 ]. 更重要的是,尽管在轻度至中度认知障碍期间进行了抗Aβ免疫接种,但所有患者都表现出持续的进行性痴呆,最终死于AD[ 29 ]. 此外,Aβ免疫治疗临床试验也充满了挫折。 在6%的免疫人群发生无菌性脑膜脑炎后,AN-1792临床试验暂停[ 32 ]. 最近,巴匹纽珠单抗被动免疫治疗临床试验因一些携带载脂蛋白(ApoE)的个体出现脑血管源性水肿而变得复杂 ε4 等位基因,迫使这些受试者退出试验[ 33 ]. 尽管有这些不良事件,最近对AD AN-1792免疫个体的分析显示,神经突和τ病理学明显减少[ 34 , 35 ].
在这篇文章中,我们报告了在9名临床诊断为AD的患者中观察到的神经病理学和生物化学变化,这些患者是在AN-1792标签下积极免疫原纤维aβ1-42的。 我们将目前的结果与我们之前对2例AN-1792免疫病例的调查进行了比较[ 28 ]以及来自6例未经治疗的AD病例和5例非痴呆对照(NDC)个体的数据。
方法 人类受试者 这项研究涉及9名临床诊断为AD的患者,他们积极接种了Aβ肽1-42(AN-1792)。 其中6例由南安普顿大学医学院(USSM)的J.Nicoll博士提供,3例由加利福尼亚大学圣地亚哥分校(UCSD)的E.Masliah博士提供(表 1 ). 为了进行比较,Banner Sun Health Research Institute(BSHRI)的T.Beach博士提供了6例非免疫性AD病例和5例NDC病例。 在免疫组(n=9)中,平均患者死亡年龄为81岁,2名女性,7名男性,平均病程为8年。 在NDC患者(病例#1-5)中,平均死亡年龄为82岁(2名女性和3名男性),而在AD患者(病例#6-11)中,死亡年龄平均为82岁,平均病程为9年(5名女性和1名男性)。 表中给出了他们的个人年龄、性别、ApoE基因型、疾病持续时间、免疫原剂量、注射次数和抗体滴度以及首次免疫剂量应用后的存活时间(如适用) 1 在9名临床诊断为AD的免疫个体中,经尸检,有2例被归类为非AD痴呆病例(USSM的21号病例和UCSD的22号病例),其神经病理学诊断分别为进行性核上性麻痹(PSP)和海马硬化(HS)伴严重额叶萎缩。 1例(#19)经神经病理学诊断为AD的路易体变异。
表1。
案例ID 过期年龄(年) 性别 ApoE基因型 疾病持续时间(年) Imm公司。 剂量(μg) #注射次数 平均抗体反应(ELISA单位) PIST(月)
国家数据中心
1-BSHRI公司 77 F类 2/3 - - - - -
2-BSHRI公司 81 F类 4/4 - - - - -
3-BSHRI公司 83 M(M) 3/4 - - - - -
4-BSHRI公司 88 M(M) 2/3 - - - - -
5-BSHRI公司 81 M(M) 3/3 - - - - -
平均值 82
AD公司
6-BSHRI公司 76 F类 4/4 6 - - - -
7-BSHRI公司 82 M(M) 第3页,共4页 7 - - - -
8-BSHRI公司 86 F类 3/4 5 - - - -
9-BSHRI公司 80 F类 3/3 12 - - - -
10-BSHRI公司 85 F类 3/3 9 - - - -
11-BSHRI公司 81 F类 第4页,共4页 15 - - - -
平均值 82 9
免疫性AD*
12-USSM公司 71 F类 3/4 10 225 8 1:4072 44
13-苏联 81 M(M) 不适用 7 50 8 1:1707 57
14-苏联 82 M(M) 3/4 6 50 8 1:4374分 60
15-苏联 81 M(M) 4/4 11 225 7 1:491 63
16-苏联 88 F类 3/3 11 50 7 1:137 86
19-加州大学圣地亚哥分校 78 M(M) 3/4 5 50 1 不适用 36
20-UCSD(加州大学圣地亚哥分校) 86 M(M) 3/4 9 50 1 不适用 60
平均值 81 8 6 58
免疫非ADD*
21-苏联 PSP公司 79 M(M) 2/3 6 50 8 < 1:100 51
22-加州大学圣地亚哥分校 高速 80 M(M) 3/3 5 50 1 不适用 48
形态学评估 对BSHRI AD和NDC病例进行斑块总评分、斑块密度、NFT总评分、建立阿尔茨海默病注册协会(CERAD)、神经炎斑块评分、Braak分期、WMR总评分和CAA总评分的评估(见表 2 ). 这些神经病理学参数的详细评估在以前的出版物中给出[ 36 ]. 主观上归因于免疫治疗的斑块清除率的半定量评估估计为无(0)、轻度(+)、中度(++)和广泛(++)。 通过在100 ml 10%SDS、10 mM Tris-HCl pH 7.4中溶解10立方大脑皮层组织(每侧约1 cm)来评估UCSD患者的CAA含量。 在室温下连续搅拌48小时后,剩下的唯一结构是不溶性血管簇和附着的淀粉样沉积物。 去除洗涤剂后,将容器在载玻片上风干,并用硫黄素-S染色[ 17 ].
表2。
案例ID Aβ42载荷(%) 牙菌斑清除 脑重(g) WMR总分 斑块总得分 菌斑密度 NFT总分 Cerad NP公司 制动阶段 CAA总分
国家数据中心
1 - - 950 不适用 6 零 2 非AD 我 不适用
2 - - 1275 0 7.6 稀疏的 3 非AD 二 2
三 - - 1385 0 1 零 1 非AD 我 0
4 - - 1260 4 6.8 适度的 4.3 可能的AD 三 三
5 - - 1190 0 12.5 稀疏的 6.4 非AD 三 5
AD公司
6 - - 1100 0 13.5 适度的 8.8 可能AD 四、 4
7 - - 1145 0 13 频繁的 13 确定AD V(V) 5
8 - - 1055 不适用 13.25 频繁的 8.75 确定AD V(V) 4
9 - - 765 9 12.5 频繁的 14.5 确定AD 不及物动词 1
10 - - 960 0 12.25 频繁的 12 确定AD V(V) 0
11 - - 970 10 13.25 频繁的 15 确定AD 不及物动词 10
免疫性AD*
12 4.68 + 不适用 不适用 不适用 不适用 不适用 不适用 不及物动词 不适用
13 3.32 ++ 1120 不适用 不适用 不适用 不适用 不适用 不及物动词 不适用
14 0.05 +++ 1200 不适用 不适用 不适用 不适用 不适用 不及物动词 不适用
15 2.71 ++ 不适用 不适用 不适用 不适用 不适用 不适用 不及物动词 不适用
16 2.99 + 不适用 不适用 不适用 不适用 不适用 不适用 不及物动词 不适用
19 不适用 不适用 1208 不适用 不适用 不适用 不适用 不适用 三 不适用
20 不适用 不适用 1162 不适用 不适用 不适用 不适用 不适用 四、 不适用
免疫非ADD*
21磅/平方英寸 0.75 0 不适用 不适用 不适用 不适用 不适用 不适用 不适用 不适用
22小时 不适用 不适用 1280 不适用 不适用 不适用 不适用 不适用 不适用 不适用
来自USSM的Aβ42免疫接种对阿尔茨海默病患者#12-15的临床、神经病理学和长期影响已在其他地方详细描述[ 29 , 30 , 35 ]. UCSD病例#19、20和22的神经病理学报告总结见表 三 .
表3。 UCSD AN-1792免疫病例临床神经病理分析总结
案例ID
有福了 [ 63 ]
MMSE公司 牙菌斑总数 神经炎斑块 缠结 CAA公司 神经病理学诊断
19 26 8 23 7 0 三 AD路易体变体
20 17 18 50 0 0 2 AD公司
22 28 10 4 三 0 0 HS,严重额叶萎缩
ELISA法测定可溶性和不溶性Aβ 所有步骤均在4°C下进行。 用特氟隆组织研磨机将灰质组织(100 mg)均匀化,其中6体积为20 mM Tris-HCl,5 mM EDTA,pH 7.8,含蛋白酶抑制剂鸡尾酒(PIC,罗氏诊断公司,德国曼海姆)。 匀浆以435000× 克 在使用120.2转子的Optima TLA超离心机中保持20分钟(加利福尼亚州富勒顿贝克曼)。 上清液保存为可溶性Aβ组分和总蛋白,通过Pierce BCA蛋白质分析进行定量(Rockford,IL)。 在3 ml 90%的玻璃蒸馏甲酸(GDFA)中均匀化每四毫克灰质和白质组织,并在250000× 克 在使用SW41转子(贝克曼)的贝克曼LE-80K超离心机中,在4°C下保持20分钟。 仔细收集上清液,避开顶部脂肪层。 标本在90%GDFA中均质,目的是完全溶解所有形式的Aβ(纤维、弥散、膜结合、细胞内和细胞外寡聚物)。 高速离心可以去除所有脂质,包括被GDFA完全破坏的膜相关形式,并在离心管顶部形成紧密的聚集物。 每个病例的全部体积在80%GDFA流动相中进行快速蛋白液相色谱(FPLC)尺寸排除Superose-12色谱(见下文)。 从每次运行中收集并汇集Aβ肽组分,并通过真空离心将其减少至2 ml(SpeedVac;Savant/Thermo,Waltham,MA)。 为了去除酸,每例患者在1000 MW切断管中进行透析,每次换2次水(每次1小时),换2次0.1 M碳酸氢铵溶液(每次1 h)。 样品被冻干和冻干。 然后用500μl的5 M盐酸胍(GHCl)在50 mM Tris-HCl(pH 8.0)中制备,并在4°C下摇晃过夜,重新配制样品。 总蛋白通过Pierce BCA蛋白测定进行定量。 用于定量Aβ40和Aβ42的ELISA试剂盒分别从Invitrogen(加利福尼亚州卡尔斯巴德)和Innogenetics(比利时根特)获得,并按照制造商的说明进行操作。
ELISA法测定肿瘤坏死因子-α(TNF-α) 所有步骤均在4°C下进行,并遵循先前发布的方案[ 37 ]. 使用Omni-TH电动组织研磨机在20体积的20 mM HEPES、1.5 mM EDTA、pH 7.4的PIC(Roche)中对灰质(100 mg)进行均质,并在3000× 克 在IEC Centra CL3R离心机(Thermo,Waltham,MA)中保持15分钟。 然后收集上清液并以40000× 克 使用50.4 Ti转子(Beckman)和Optima LE-80K超离心机(Beck曼)持续1小时。 再次收集上清液,并用BCA蛋白测定法(Pierce)测定总蛋白。 根据制造商的说明,使用PromoKine(德国海德堡)的试剂盒测量人类TNF-α水平。
Western Blot分析 如前所述进行蛋白质印迹[ 38 ]. 简言之,将灰质在含有PIC(Roche)的RIPA缓冲液(Sigma,St.Louis,MO)中匀浆。 离心匀浆,收集上清液,用BCA蛋白检测试剂盒(Pierce)定量总蛋白。 用SDS-PAGE分离样品(每道装载25μg总蛋白),然后电泳转移到0.45μm孔的硝化纤维素膜(Invitrogen)上,用5%的非脂肪乳在磷酸盐缓冲液(PBS)中封闭,0.5%吐温20(弗卢卡,圣路易斯,密苏里州)。 实验中使用的主要抗体包括22C11(识别APP的66-81个氨基酸;Millipore,Billerica,MA)、CT9APP(识别APP-的最后9个C末端氨基酸;Milliepore)和anti-au HT7(识别氨基酸159-163;Pierce)。 所用的二级抗体为山羊抗鼠IgG结合辣根过氧化物酶(HRP;22C11和antiau)或Pierce的山羊抗兔IgG偶联HRP(CT9APP)。 用SuperSignal WestPico化学发光底物(Pierce)、CL-X胶卷(Pierse)和柯达GBX显影剂和定影剂(Sigma)检测蛋白质信号。 使用GS-800校准密度计(Bio-Rad,Hercules,CA)和Quantity One软件(Bio-Read)进行分析。 用Restore™Western Blot Stripping Buffer(Pierce)剥离所有膜,在PBS中清洗,然后重新封闭。 使用抗鼠或抗兔肌动蛋白抗体(Abcam,Cambridge,MA)对总蛋白标准化的印迹进行再验证。
快速蛋白质液相色谱法 将大脑皮层(~3 g)切碎,放入18 ml 90%GDFA中均质,在室温下静置15 min,在240000× 克 在4°C下保持1小时。 小心地清除收集在试管顶部的分离脂质层,并丢弃试管底部的小颗粒。 将中间上清液分为500μl份,并在-80°C下冷冻。 使用Superose 12色谱柱(10×300 mm,General Electric,Uppsala,Sweden)将每等分样品提交给大小不限的FPLC,并用80%的GDFA进行平衡。 使用80%GDFA在室温下以15 ml/h的流速开发色谱,并在280 nm下进行监测。 在50-66分钟之间洗脱的部分,含有2-8 kDa M 第页 收集分子,并通过真空离心(Savant Instruments Inc)将其还原至约50μl,并储存在-80°C下。
高效液相色谱法(HPLC) 使用C8柱(4.6×250 mm,Zorbax SB,Mac Mod),使用含0.1%三氟乙酸(TFA)的0-60%水-乙腈浓度的线性梯度,在80°C下以1 ml/min的流速在120 min内展开,通过反相HPLC分离2-8 kDa FPLC组分。 在214nm处监测吸光度,共收集9个组分,并通过真空离心法缩小体积。 为了消除酸,用三次水(每次200μl)清洗试样,并用真空离心法缩小体积。 最后一次清洗后,体积减小,样品在含有50 mM二硫苏糖醇的2xLDS样品加载缓冲液(Invitrogen)中重新溶解。 如上所述,以抗Aβ40、抗Aβ42(Invitrogen)和CT9APP(Millipore)作为一级抗体,以山羊α-兔IgG HRP作为二级抗体进行Western blot。
表面增强激光解吸/电离飞行时间质谱(SELDI-TOF)质谱(MS)。 Aβ40/42方法 使用Western blot分析并发现含有Aβ的HPLC峰,按照之前发布的方案进行SELDI-TOF MS[ 39 ]. 简言之,将捕获抗体、抗Aβ40和抗Aβ42抗体(Invitrogen)以0.38 mg/ml的浓度加载到PS20蛋白质芯片阵列(Bio-Rad,Hercules,CA)上。然后应用HPLC Aβ峰或Aβ1-40或1-42肽标准(阳性对照)。 分子质量赋值是由Bio-Rad SELDI蛋白质生物学系统II中的100个平均注射结果产生的,使用蛋白质芯片肽质量校准试剂盒(Bio-Rad)进行外部校准。
SELDI-TOF MS,6E10方法,蛋白质芯片β-淀粉样蛋白MPD试剂盒 将含有Aβ的HPLC峰汇集在一起,得到每例一个合并样品。 所有步骤均在室温下进行。 根据制造商的说明(Bio-Rad)制备内部标准品和校准品。 对于每个HPLC和校准样品(Aβ肽:1-16、1-38、1-40和1-42),50μl/ml内标物AβCys1-24(M 第页 =2979.3)。 蛋白质芯片阵列上的每个点用5μl PBS平衡5分钟,然后装入5μl样品或校准剂,并在加湿室中培养1小时。取出样品/校准剂,在15 ml锥形管中用10 ml洗涤缓冲液(PBS,0.5%Triton X-100)洗涤每个阵列3次 每次5分钟,然后在PBS中3次,每次5分钟。 为了淡化阵列,每个芯片在10 ml 0.1 M HEPES中清洗5分钟,然后风干。 向5 mgα-氰基-4-羟基肉桂酸(CHCA)中加入200μl乙腈和200μl 1.0%TFA。 将溶液涡流2分钟,然后以1000× 克 持续1分钟以清除微粒。 将饱和CHCA制成20%的CHCA溶液(以乙腈和1.0%TFA的1:1比例稀释),并旋转1分钟。将20%CHCA溶液施加于每个点(1μl)并风干。 如上所述进行分子质量分配和校准。
结果 一、临床和神经病理观察 我们检查了9名临床诊断为AD的患者,他们接受了AN-1792免疫治疗。 在使用SDS去除脑实质后,对19、20和22号病例的皮质血管进行整装准备,发现存在不溶性CAA(图 1 ). 比较参考,表 2 显示了在5个NDC个体(病例#1-5)和6个未免疫的AD患者(病例#6-11)中观察到的神经病理学参数。 以前的出版物中描述了5例来源于USSM的免疫病例(病例#12-16)中观察到的神经病理学变化,这些免疫病例用AN-1792抗原治疗[ 29 , 30 , 35 ]. 表中给出了他们的Aβ42负荷和斑块清除和Braak阶段的半定量估计程度 2 这组个体分别接受7-8次抗原注射,每次剂量为50或225μg,初始免疫后平均存活时间为62个月(范围44-86个月)(表 1 ). 其中四名患者进行了ApoE基因分型,其中三人携带 ApoEε4 等位基因。
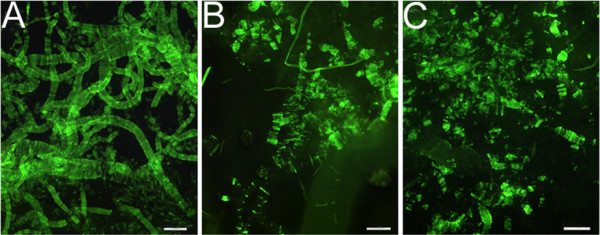
图1。
使用大脑皮层SDS裂解物评估UCSD患者CAA的血管硫黄素S .A)19号病例显示严重CAA。 B) 病例20显示中度CAA。 C) 病例22表现为中度CAA,尽管该病例的神经病理学报告为无CAA(表3)。 这些差异可能是由于不同的方法评估或采样地点造成的。 比例尺=250μm。
临床诊断为AD的USSM病例#21接受了6次免疫接种,每次50μg(表 1 ). 然而,在神经病理学检查中,该病例被重新归类为PSP。 因此,Aβ42负荷非常低(0.75%)(表 2 )ELISA测定的可溶性和不溶性Aβ水平也可以忽略不计(表 4 ).
表4。
总经理 GM GDFA/GHCl可溶性Aβ WM-GDFA/GHCl-可溶性Aβ GM三溶性Aββ
TNF-αpg/mg总蛋白* Aβ40 ng/mg总蛋白 Aβ42 ng/mg总蛋白**# 总Aβng/mg总蛋白*** Aβ40 pg/mg总蛋白 Aβ42 pg/mg总蛋白 总Aβpg/mg总蛋白 Aβ40 pg/mg总蛋白 Aβ42 pg/mg总蛋白 总Aβpg/mg总蛋白
国家数据中心
1 24 15 488 503 102 454 556 0 0 0
2 13 82 911 993 82 428 510 166 438 604
三 14 4 105 109 83 429 512 0 0 0
4 12 56 920 976 66 570 636 0 0 0
5 17 167 2168 2335 81 2596 2677 0 142 142
平均值 16 65 918 983 87 836 923 28 97 124
AD公司
6 11 5405 8152 13557 101 4122 4223 116 138 254
7 13 374 5910 6284 923 2379 3302 0 88 88
8 13 179 4773 4952 90 5156 5246 148 93 241
9 13 394 2664 3058 136 965 1101 254 66 320
10 15 96 5344 5440 91 1931 2022 0 35 35
11 18 7169 2847 10016 8940 13669 22609 151 0 151
平均值 14 2269 4948 7218 1714 4704 6417 112 70 182
免疫性AD
12 45 6347 58 6405 - - - 1260 0 1260
13 37 9925 1226 11151 - - - 13169 262 13431
14 32 252 31 283 - - - 0 0 0
15 32 1067 421 1488 - - - 629 26 655
16 43 8568 1570 10138 - - - 7556 142 7698
19 33 764 1565 2329 661 2141 2802 498 0 498
20 34 6315 7696 14011 7363 29669 37032 565 160 725
平均值 34 4748 1795 6544 4012 15905 19917 3382 84 3467
免疫非ADD
21磅/平方英寸 30 1 18 19 - - - 0 0 0
22小时 13 三 333 336 158 1245 1403 0 0 0
表 三 介绍了UCSD提供的三例患者的临床和神经病理学数据。 经尸检神经病学检查,19号和20号病例分别被认定为AD和AD的路易体变体。 临床诊断为AD的22号免疫患者在神经病理学检查中发现为HS病例,与免疫AD组的平均水平相比,其aβ水平也较低(表 4 ). 所有三个人只接受了单剂量的AN-1792抗原。
二、。 AβELISA定量 评估免疫治疗效果的关键是通过ELISA对Aβ40、Aβ42和总Aβ肽进行定量,即在灰质(GM)和白质(WM)中以Tris-soluble和GDFA/GHCl-soluble形式存在。 USSM没有冷冻WM用于ELISA分析。 这些值如表所示 4 为了进行比较,表中还列出了在未免疫的AD和NDC个体中观察到的类似值 4 GM GDFA/GHCl Aβ42和总AβGM GDFA-GHCl水平在GM和WM组织分区中观察到NDC和AD人群之间的Aβ水平存在显著差异(分别为p=0.0026和0.0066,未配对,双尾t检验)。
免疫AD病例GM中总Aβ水平的总体变异程度很大,GDFA/GHCl可溶性物种的总蛋白为283至14011 ng/mg(平均值=6544),对应的Tris-soluble Aβ组分的总蛋白则为0至13431 pg/mg(均数=3467)(表 4 ). 如受试者#13和#14所示,免疫治疗的最终结果存在显著差异。 两人都接受了8次50μg AN-1792注射(表 1 ). 在后一种情况下,组织学观察[ 29 , 30 ]通过ELISA定量进行匹配,结果显示可溶性和不溶性Aβ肽总量相对较少,而病例#13的GDFA/GHCl可溶性Aβ水平第二高,Tris可溶性Aβ水平最高。 相反,与免疫组相比,病例#14在两种溶剂中的Aβ水平最低。 在这两个人中,自第一次免疫以来的存活时间几乎相同:57个月和60个月,这表明Aβ淀粉样斑块病理学的存在与否显然与AD痴呆的进展和致命结局无关。
免疫组和非免疫组AD组之间平均Aβ水平的比较也表明可溶性和不溶性Aβ含量的总体差异很大(表 4 ). 免疫和非免疫个体之间的GDFA/GHCl可溶性aβ42存在统计显著差异(p=0.039),前者较低。 另一方面,在免疫队列中,Aβ40的水平反常增加,几乎是非免疫AD对照组中观察到的平均数量的两倍,尽管差异没有达到统计学意义。 在一名免疫个体(病例#14)中,总Tris-soluble Aβ水平低于检测限。 其余6名免疫个体的平均值是未免疫AD病例中观察到的平均值的22倍。
鉴于NDC、AD和Aβ免疫AD患者的病理变化程度很高,如表所示 4 ,同样重要的是要考虑单独权衡数据,而不是只使用组平均值。 例如,在NDC组中,病例#5的GM和WM GDFA/GHCl可溶性Aβ水平异常高,因此,该组的平均值向上倾斜,比NDC组其他成员的平均值大4-5倍。 因此,该病例应归类为非痴呆型“高度病理控制”。 在AD组中,11号个体WM中的GDFA/GHCl可溶性Aβ异常高,再次将平均值向上倾斜,这是该组其他人观察到的平均值的7倍。 在免疫组中,病例#20的WM-GDFA/GHCl-可溶性总Aβ水平是病例#19观察值的13倍。 在Tris-soluble Aβ组分中,免疫的AD个体#13和#16的Aβ分别是该组其余5个个体的21倍和12倍。 这些升高的值也反映在GM GDFA/GHCl可溶性aβ组分中,虽然程度较低,但只与中度和轻度斑块清除相关[ 29 , 30 ]. 狄克逊Q检验[ 40 ]也将这些个体识别为异常值。
有趣的是,7例免疫性神经病理学确诊的AD病例中,有3例的总GM GDFA/GHCl可溶性aβ值大大低于6例非免疫性AD病例的平均值(总蛋白7218 ng/mg),这表明抗体可能已清除了这些病例中的aβ。 其余4例免疫病例的值等于或高于未免疫AD组的平均值。
如上所述,在接受UCSD提供的AN-1792免疫原治疗的3例患者中,有一例神经病理学诊断为HS。 其余2名患者,即19号和20号病例,表现出AD的神经病理学,如表所示 三 从Aβ免疫分析的角度来看,病例19具有相对中等水平的GM和WM GDFA/GHCl可溶性Aβ肽以及Tris-可溶性Aβ。 相比之下,病例#20显示了20例受调查病例中GM和WM-GDFA/GHCl-可溶性Aβ肽的最高水平,包括作为对照的5例NDC和6例非免疫性AD病例。
此外,抗体反应也存在高度的变异性(表 1 ). 例如,病例12和病例14的抗体滴度最高,但从神经病理学角度来看,病例12几乎没有斑块清除的证据,而病例14有广泛的aβ清除。 AβELISA数据也显示了病例#12和病例#14的类似模式(表 4 ). 病例15的抗体滴度相对较低,但aβ清除率适中(表 2 ). 21号病例的抗体滴度较低,这与免疫背景无关,因为这是一例PSP患者。
三、 载脂蛋白E基因型 神经病理学诊断为AD的七分之六的患者进行了ApoE基因分型。 其中5人携带ε4等位基因(表 1 ). ApoE状态与GDFA/GHCl提取的GM Aβ总量或Tris可溶性组分中存在的GM Aβ总量之间没有直接相关性(表 4 ).
四、 肿瘤坏死因子-αELISA定量 肿瘤坏死因子-α是一种促炎性细胞因子,与非免疫性AD患者的平均水平相比,在免疫性AD个体中平均升高了2.4倍(总蛋白34 pg/mg,总蛋白14 pg/mg;p<0.0001,表 4 ). 值得注意的是,即使在Aβ(#14)减少最显著的免疫性AD患者中,TNF-α水平仍高于非免疫性AD组(表 4 ).
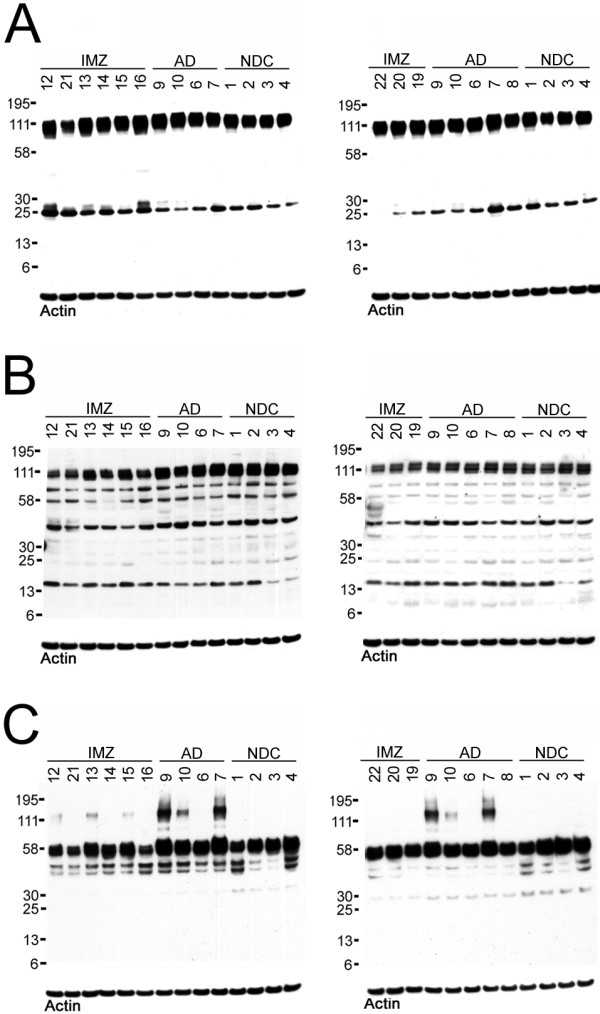
V.灰质匀浆的蛋白质印迹 9例免疫病例的大脑皮层在RIPA缓冲液中直接均质,并使用22C11、CT9APP和tau HT7抗体进行Western blots分析。 为了进行比较,还纳入了4-5例AD和4例NDC病例。 与未免疫的AD和NDC个体相比,N末端定向抗体22C11在免疫病例中的总APP无显著差异(图 2安培 ). 然而,25 kDa APP N末端肽的量有一些波动(图 2安培 ). CT9APP抗体在~13 kDa处检测到CT99/83,在~40 kDa时检测到一条带,这条带存在于所有研究样本中(图 2B型 ). 这种由CT9APP抗体揭示的肽很有意思,因为在散发性AD和NDC病例的PS突变中也存在类似大小的条带[ 38 ]以及LaFerla等人设计的三重转基因小鼠(3XTg)(A.Roher,未发表的观察结果)。 需要进一步的研究来阐明这种假定更长的APP C末端肽及其蛋白水解产物的性质。 免疫组与非免疫组之间CT99(~13kDa带)水平无差异。 25和40 kDa带可能代表来自未修饰APP分子的互补N端和C端物种。 CT9APP抗体还显示,与未免疫的AD和NDC组相比,免疫病例中总APP的量适度减少,尽管22C11没有显示出这种趋势。 该抗体在~58和~75 kDa处还显示了另外两条APP C末端相关带(图 2B型 ). 这些观察结果与22C11和CT9APP可以与淀粉样前体样蛋白(APLP)交叉反应的警告一致。 Western blot HT7 tau抗体模式的总体比较表明,AD免疫和非免疫样品之间没有显著差异,但表现出抗SDS二聚体形式tau的7号和9号AD病例除外(图 2摄氏度 ).
图2。
转基因匀浆的蛋白印迹 每个通道中总共装载了25μg蛋白质。 A) 22C11对抗APP的氨基酸残基66-81,B)CT9APP对抗APP最后9个氨基酸残基,C)tau HT7对抗tau的氨基酸残基159-163。 有关更多详细信息,请参阅结果部分。 免疫IMZ; 阿尔茨海默病; NDC,非痴呆控制。
六、 柱色谱法 通过FPLC初步分离后,含有Aβ肽的组分在50-62分钟内洗脱(相当于12.5-15.5 ml洗脱溶剂),通过C8反相HPLC分离(图 三 ). 对3例免疫病例进行了Western blot生化鉴定。 案例19相对于aβ40亚型,主要是二聚aβ42肽的数量减少(图 3A级 ). 另一方面,AN-1792病例#20具有等摩尔水平的Aβ40和Aβ42肽(图 第3页 ). HS病例#22含有极少量的Aβ40和更丰富的Aβ42相关肽补体。 用CT9APP抗体探测的蛋白质印迹证明在图中约13 kDa处存在预期的CT99和CT83 APP C末端片段 3C公司 此外,它们还显示了一条带有M的肽带 第页 约40 kDa。
图3。
HPLC分离UCSD免疫病例的灰质(C8反相) 图中显示了使用抗Aβ40、抗Aβ42和CT9APP抗体开发的Western blotting研究的HPLC色谱部分。 对角线表示乙腈梯度。 泳道10含有标准Aβ40或Aβ42合成肽。 A) 和B),分别对应于神经病理学确诊的19号和20号AD病例。 C) 对应于神经病理学证实的海马硬化。 m、 Aβ单体; d、 Aβ二聚体; t、 β三聚体。
七、。 质谱法 在之前的一项研究中,我们通过基质辅助激光解吸/电离飞行时间质谱法(MALDI-TOF MS)鉴定了大量肽,该研究涉及用AN-1792免疫的2名AD患者大脑中残留的aβ相关肽。 反射子MALDI-TOF在其单同位素M中证实了这些肽的存在 第页 形式[ 28 ]. 在本次调查中,9例受试者中有3例(UCSD病例#20-22)采用质谱法进行了调查。 以Aβ40、Aβ42和6E10为捕获抗体,采用SELDI-TOF MS鉴定Aβ相关肽。 一组Aβ相关肽被鉴定为N末端起始于1、2、3-焦谷氨酰(3pE)、4、5、6、11和17,C末端终止于40、42、43、46、47、49、50、61、62、68、73、82、96和99(Aβ相关氨基酸序列中给出的数据,其中残基99对应于APP的残基695 695 分子,见表 5 ). 观察到Aβ2-40和1-43的二聚体以及鉴定为3pE-40+4-40和3pE-40+2-40的二聚Aβ杂交体。 可以理解,有几个较长的Aβ相关肽覆盖APP跨膜结构域以外的氨基酸序列。 我们的研究揭示了分泌酶在β-位点1和11、α-位点17以及推定的γ-位点40、42、43、46、47、49和50处的切割。 发现从残基2、3、4、5和6开始的N末端Aβ序列较短,可能是由于氨肽酶活性所致,这在所有AD病例中都常见[ 41 ]. 翻译后修饰,如残基3戊二酰环化为焦戊二酰[ 42 ]还观察到Met的氧化和Ser和Thr的无规甲酰化。 后一种修饰可能是样本暴露在甲酸中的伪影。 虽然由于多肽的复杂混合物阻碍了激光电离速度,因此很难通过质谱对多肽进行量化,但正如我们的HPLC/Western blot研究所示,在AD中,二聚体aβ相关低聚物相对于单体的数量具有很强的优势(见图 三 )以及我们实验室进行的其他研究表明,二聚体Aβ是最稳定和最丰富的寡聚体结合[ 43 - 45 ].
表5。 用Aβ40、Aβ42抗体和6E10捕获的SELDI-TOF肽
M(M) 第页 观察 M(M) 第页 计算 肽序列
3371.2 3373 17-49(2f)
3442.2 3444.1 17-50(f)
3486.1 3488.1 17-50(牛/2f)
4514.5 4514.1 1-42
4925.5 4924.7 2-47
4940.7 4942.6 1-46(牛)
5371.4 5670.8 11-62
6178.3 6177.2 6-61(3f)
6723.3 6724.9 11-73(牛)
7178.2 7180.3 4-68(3箱)
8202 8204.1 3pE-40+4-40(4箱)
8411.7 8412.3 3pE-40+2-40(牛/2f)
8450.4 8445.4 二聚体2-40(ox)
8565.4 8563.9 5-82
9265.5 9262.4 二聚体1-43(2ox)
9273 9273.7 17-99(4箱)
9624 9623.1 11-96(公牛)
讨论 在APP转基因小鼠模型和AD患者中,主动和被动Aβ免疫的结果与有效的淀粉样斑块破坏一致,尽管在接受治疗的啮齿动物和人类中,老年核心消退的程度是高度可变的。 目前,约10000名受试者正在积极进行40多项Aβ免疫治疗临床试验(阿尔茨海默病研究论坛; http://www.alzforum.org/new/detail.asp?id=2409 ; 2010年4月2日)。 有趣的是,在第二阶段临床试验中,Bapineuzumab被动免疫治疗在两个主要结果指标(ADAS-cog和痴呆症残疾评估)中没有统计学意义,尽管 事后(post-hoc) 对少数完美主义者的探索性分析显示出一些小的治疗效果[ 33 , 46 ]. 然而,抗淀粉样蛋白免疫疗法是否会显著改善AD患者的认知能力和生活质量,甚至阻止痴呆的进展,还有待确定。
本研究中发现的潜在混淆临床误诊比例充分说明了迫切需要可靠的AD生物标记物。 在9名临床诊断为AD痴呆的患者中,有2名随后被神经病理学认定为PSP和HS病例。 有趣的是,从我们自己的BSHRI脑库中观察到的结果表明,只有大约50%的AD病例应被视为神经病理学上的简单AD[ 36 ]. 其余代表AD病理学与其他神经退行性疾病相结合,导致严重的认知和智力退化,如血管性痴呆、路易体痴呆、额颞叶痴呆、帕金森氏病、PSP、HS或无特殊病理学的痴呆。 由于缺乏对AD的可靠临床诊断,并且在接受免疫治疗的许多尸检患者中几乎或完全没有淀粉样斑块,因此无法确定所有这些患者在治疗开始时是否确实表现出AD斑块病理,或者是否有发展成这种病理的风险。 介绍检测淀粉样斑块负荷的成像技术,例如基于对比剂Aβ结合染料化合物的成像技术(以匹兹堡化合物-1为例)( 11 C-PiB-PET)将有助于选择适合免疫治疗的候选人。
我们的研究小组和其他人员对AN-1792临床试验参与者进行了详细的尸检[ 24 - 31 ]. 一个关键的观察结果是,免疫分析研究显示,接受治疗的个体对免疫疗法的反应范围很广,这反映在Aβ肽水平的极端可变性上。 假设治疗开始于有完整淀粉样斑块的患者,神经病学检查始终显示局限于老年斑块明显受损的广泛区域。 此外,ELISA分析表明,在某些情况下,Aβ的总量因治疗而减少。 这种患者间的变异性表明,有必要对治疗方案进行个性化,并精确滴定剂量,以达到绝经后的疗效和安全性。
我们以前的报告[ 28 ]检测了2例用AN-1792抗原免疫的AD病例。 其中一例患者接受了2次225μg免疫原肌肉注射。 第二次免疫9个月后,患者发生非终末无菌性脑膜脑炎。 在第二个病例中,给药3剂225μg,患者在第一次免疫后一年因“发育不良”而死亡。 两人都携带 ApoEε3/ε4 基因型。 在这两例患者中,尸检显示,大脑的某些区域出现了几乎完全明显的淀粉样斑块破裂,伴有溶解,但没有Aβ从大脑中清除。 血管和弥漫性淀粉样沉积物都抵抗AN-1792破坏。 发生脑膜脑炎的患者中GHCl-可溶性Aβ的总量为868μg/g湿重,而第二名患者为186μg/g干重。 非免疫AD(n=31)和NDC(n=22)的平均Aβ值分别为406μg/g湿重和221μg/g干重[ 28 ]. 然而,虽然总体趋势可以等同,但这些值与本研究中的值并不直接可比,因为Aβ提取技术不同,所报告的平均值范围也不同,以总蛋白ng/mg与湿重μg/g表示。 在本研究中,两例患者的总Aβ肽水平差异很大。 与第二例相比,脑膜炎患者的三溶性Aβ高4.5倍,GHCl WM Aβ高5.7倍。 在前一篇论文中的两个免疫病例中,Tris-soluble Aβ的水平高于NDC和非免疫AD组的水平[ 28 ]. 在当前的研究中也出现了同样的趋势,AD免疫病例(病例#14除外)的Tris-soluble Aβ水平高于AD和NDC人群的平均水平。 在我们之前的研究中,通过硫黄素-S染色评估,血管淀粉样蛋白沉积量也明显显著增加[ 28 ].
事实上,淀粉样斑块的消除并没有改变痴呆症的下降轨迹[ 29 ]令人失望,这表明淀粉样斑块本身并不是痴呆症的直接潜在原因。 最近的几项研究表明,经过两年的随访, 11 AD患者C-PiB摄取量保持不变[ 47 , 48 ]. 然而,虽然淀粉样蛋白沉积稳定,但显著减少(约20%, 对 =0.01)区域葡萄糖代谢率[ 48 ]. 此外,串行 11 NDC、轻度认知障碍(MCI)和AD患者的C-PiB-PET和MRI显示,淀粉样变本身不足以导致认知功能下降,而认知功能下降似乎是由脑萎缩和神经变性引起的[ 49 ]. 有趣的是,随着时间的推移,NDC(1.3毫升/年)、MCI(2.5毫升/年)和AD(7.7毫升/年)的心室扩张量继续增加,这是自然衰老或脑病理相关萎缩的表现[ 49 ]. 淀粉样斑块虽然无疑是有害的,但可能是大脑管理aβ积聚的一个救援计划[ 50 ]. 一些研究表明,可溶性二聚体Aβ物种代表了这些分子中毒性最强的形式[ 44 , 51 ]这意味着只关注淀粉样斑块修复过于有限。
尽管令人失望的是,淀粉样斑块的破坏既不能治愈痴呆,也不能阻止痴呆的进展,但现在宣布这一策略失败还为时过早。 首先,淀粉样斑块是主要痴呆病理学的假设尚未得到严格的检验。 生化解剖显示淀粉样斑块多于Aβ堆积,但实际上代表复杂的多分子组合[ 52 , 53 ]. 尸检显示,免疫治疗并不能完全逆转某些淀粉样斑块[ 28 ]和残余物,被称为“崩塌”斑块[ 25 ]或“被虫蛀”的斑块[ 27 ]由不溶性分子组成的物质持续存在。 尽管免疫治疗具有令人印象深刻的形态学效果,但“淀粉样斑块骨骼残留物”可能含有有毒成分,并继续对痴呆症的发展产生有害的遗留影响。 此外,构成斑块沉积物的Aβ谱在结构上可能具有足够的多样性,以阻止迄今为止使用的免疫治疗药物的完全破坏。 识别Aβ分子N末端的人源化单克隆抗体[ 46 ]将无法识别已证明在人类老年斑中普遍存在的终末期Aβ物种[ 41 ]. 此外,弥漫性斑块周围缺乏反应性小胶质细胞,无法引发炎症反应[ 54 , 55 ]. 这种现象可能是由于缺乏与小胶质细胞表面糖胺聚糖结合的AβHHQK结构域所致[ 13 , 14 ]. 此外,主要由P3(Aβ残基17-42)组成的弥漫性斑块[ 56 ]),可通过直接针对AβN末端结构域的免疫治疗来避免破坏。 α-分泌酶裂解不产生淀粉样肽,因此被认为是一种良好的治疗途径。 然而,有证据支持P3激活JNK和caspase-8导致神经元凋亡的论点[ 57 ]. 除了淀粉样蛋白和弥漫性斑块沉积外,AD患者还存在NFT,这些病变在接受抗Aβ免疫治疗的患者中持续存在[ 24 , 25 , 27 , 28 , 31 , 35 ]. 由于阿尔茨海默病患者通常同时存在几种不同类型的病理和生化损伤,因此要彻底治愈痴呆症,可能需要解决的不仅仅是淀粉样斑块。
由于进化保守的Aβ分子的基本功能尚不清楚,因此大规模、长期使用抗淀粉样斑块免疫治疗变得复杂。 一种可能性是淀粉样蛋白沉积通过在血脑屏障(BBB)被突破的地方形成一个斑块来发挥血管损伤修复功能[ 17 ]. 对转基因小鼠和人类的研究表明,Aβ免疫疗法对血管完整性和功能产生强大的,有时是有害的影响[ 30 , 31 , 58 - 60 ]. 此外,直接的临床经验证实 ApoEε4 淀粉样蛋白免疫治疗更容易对基因产生严重的不良影响。 虽然尚不清楚这些反应是血管病理学的原因还是后果,但不幸的是,它们确实表明AN-1792是完全禁止使用的,或者必须谨慎应用于已知最易患AD的患者亚群。
免疫性AD病例中TNF-α的生成增强表明,部分由抗原-抗体相互作用产生的持续炎症反应。 Aβ的作用和随后小胶质细胞通过Fc受体的摄取刺激了白细胞介素(IL)-1、IL-6、IL-10、TNF-α和巨噬细胞集落刺激因子等分子的分泌,从而促进神经炎症和BBB的开放。 在设计有效的免疫疗法时需要考虑这些自相矛盾的反应[ 61 ]. 此外,TNF-α通过减少胰岛素降解酶的表达来抑制Aβ降解[ 62 ].
尽管近十年来淀粉样蛋白斑块缓解方面取得了可喜的进展,但淀粉样蛋白假说是否代表了散发性AD病理的最终机制解释尚不明确。 尽管淀粉样斑块是导致痴呆的主要原因,但目前对抗这些病变的努力需要进一步发展。 除了检查免疫治疗后大脑中淀粉样斑块显著破裂的病理生理学基础外,外周池在增加循环Aβ水平中的作用需要一个完整的机制解释。
结论 总之,我们的结果显示,在免疫个体中,aβ40和aβ42肽的总比例存在很大差异,SDS稳定的二聚体形式通常优于单体形式。 此外,SELDI-TOF MS证明了一系列Aβ相关肽,主要向APP的C端结构域延伸,这在一定程度上可能是由于我们使用的强大提取条件,能够完全分散膜结构。 与NDC和非免疫AD组相比,免疫AD患者GM中的促炎性TNF-α水平显著增加。 β-淀粉样蛋白免疫导致淀粉样蛋白斑块破裂和清除的程度差异很大,其水平从低于检测限到超过ELISA中非免疫性AD病例的值不等。 这些特异性反应,再加上表征衰老和疾病状况的广泛生物和病理变化,将使解释治疗研究得出的数据变得困难。 此外,这种变异极大地使AD合适生物标志物的识别复杂化。 我们的数据表明,治疗结果将取决于注射免疫原的质量和数量以及患者的免疫反应,ApoE表型似乎可以调节斑块和血管淀粉样蛋白清除的有效性。 在尝试将Aβ抗体作为AD治疗工具时,应考虑到Aβ肽在人体内的所有物理化学性质。 尽管在某些情况下,AN-1792去除淀粉样斑块的效果令人印象深刻,但阿尔茨海默病的临床进展没有相应的改变。
缩写词表 11 C-PiB:匹兹堡化合物; Aβ:淀粉样蛋白β; AD:阿尔茨海默病; 载脂蛋白E; APP:淀粉样β前体蛋白; BBB:血脑屏障; BSHRI:Banner Sun健康研究所; CAA:脑淀粉样血管病; CERAD:建立阿尔茨海默病注册中心联盟; CHCA:α-氰基-4-羟基肉桂酸; F: 女性; f: 甲酰基; FLA:额叶萎缩; FPLC:快速蛋白质液相色谱法; GDFA:玻璃蒸馏甲酸; GHCl:盐酸胍; GM:灰质; 高效液相色谱法; HS:海马硬化; IL:白细胞介素; Imm:免疫; M: 雄性; MALDI-TOF:基质辅助激光解吸/电离飞行时间; MCI:轻度认知障碍; MMSE:简易精神状态检查; MS:质谱法; 不适用:不可用; NDC:非痴呆控制; NFT:神经纤维缠结; 非ADD:非阿尔茨海默病痴呆; NP:神经炎斑块; ox:氧气; pE:戊二酰; PIC:蛋白酶抑制剂鸡尾酒; PS:早老素; PSP:进行性核上性麻痹; TFA:三氟乙酸; Tg:转基因; TNF-α:肿瘤坏死因子α; USCD:加州大学圣地亚哥分校; 南安普顿大学医学院; PBS:磷酸盐缓冲盐水; RT:室温; SELDI-TOF:表面增强激光解吸/电离飞行时间; WM:白质; WMR:白质稀疏
竞争利益声明 JARN是阿尔茨海默病免疫项目的顾问/顾问:Elan Pharmaceuticals、GSK、Novartis、Roche、Janssen Alzheimer Immunotherapy Research and Development。 MNS获得BMS、Avid、GE、Bayer、Baxter、Wyeth、Janssen、Lilly和Medivation的资助(临床试验)。 MNS也是Janssen/Pfizer、Amerisciences、Eisai和GSK的顾问/咨询委员会成员,并从Ameriscences和Wiley获得版税。 TGB从AVID-Bayer GE Radiopharmatices获得资金。 其余作者没有相互竞争的兴趣。
作者的贡献 MNS和TGB对AD和NDC患者进行了临床和神经病理学评估。 JAN和EM提供了AN-1792例患者的临床和神经病理学数据。 WMK、RLP、DCL、CLM和IDD参与了数据的收集和分析。 AER、TAK和EMC参与了实验设计和手稿的最终制作。 所有作者都参与了手稿的修订和编辑。
参与者信息 Chera L Maarouf,电子邮件: chera.maarouf@bannerhealth.com。
Ian D Daugs,电子邮件: ian.daugs@bannerhealth.com。
Tyler A Kokjohn,电子邮件: tkokjo@midwestern.edu。
Walter M Kalback,电子邮件: wmkalb@att.net。
R Lyle Patton,电子邮件: pattfam@cox.net。
Dean C Luehrs,电子邮件: dluehrs@cox.net。
Eliezer Masliah,电子邮件: emasliah@ucsd.edu。
James AR Nicoll,电子邮件: J.Nicoll@soton.ac.uk。
Marwan N Sabbagh,电子邮件: marwan.sabbagh@bannerhealth.com。
Thomas G Beach,电子邮件: thomas.beach@bannerhealth.com。
Eduardo M Castaño,电子邮件: ECastano@leloir.org.ar。
Alex E Roher,电子邮件: alex.roher@bannerhealth.com。
致谢 本研究得到了美国国家老龄研究所(NIA)资助:R01 AG-19795,NIA亚利桑那州阿尔茨海默病核心中心P30 AG-19610,以及亚利桑那州向亚利桑纳州阿尔茨海默病研究联盟(Arizona Alzheimer's Research Consortium)的资助。 我们对Douglas Walker博士(BSHRI)进行ApoE基因分型表示感谢。 JARN获得了英国阿尔茨海默病研究信托基金的资助。 Clive Holmes和Delphine Boche参与了南安普顿大学医学院病例的临床和神经病理学评估。
工具书类
Brookmeyer R、Johnson E、Ziegler-Graham K、Arrighi HM预测阿尔茨海默病的全球负担。 老年痴呆症。 2007; 3:186–191. doi:10.1016/j.jalz.2007.04.381。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
2010年阿尔茨海默病事实和数字。 老年痴呆症。 2010; 6:158–194. doi:10.1016/j.jalz.2010.01.009。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Glenner GG、Wong CW。 阿尔茨海默病:一种新型脑血管淀粉样蛋白的纯化和表征的初步报告。 生物化学与生物物理研究委员会。 1984; 120:885–890. doi:10.1016/S0006-291X(84)80190-4。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Masters CL、Simms G、Weinman NA、Multhaup G、McDonald BL、Beyruther K.阿尔茨海默病和唐氏综合征中的淀粉样斑块核心蛋白。 美国国家科学院院刊,1985年; 82:4245–4249. doi:10.1073/pnas.82.12.4245。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Hardy JA,Higgins GA。阿尔茨海默病:淀粉样级联假说。 科学。 1992; 256:184–185. doi:10.1126/science.1566067。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Selkoe DJ。阿尔茨海默病:基因、蛋白质和治疗。 《生理学评论》,2001年; 81:741–766. doi:10.1152/physrev.2001.81.2741。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Joseph J、Shukitt-Hale B、Denisova NA、Martin A、Perry G、Smith MA。哥白尼重访:阿尔茨海默病中的β淀粉样蛋白。 神经生物老化。 2001; 22:131–146. doi:10.1016/S0197-4580(00)00211-6。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Obrenovich ME、Joseph JA、Atwood CS、Perry G、Smith MA。淀粉样β:大脑的(生命)保护剂。 神经生物老化。 2002; 23:1097–1099. doi:10.1016/S0197-4580(02)00038-6。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Lopez-Toledano MA,Shelanski ML.β-淀粉样肽在神经干细胞发育中的神经原性作用。 神经科学杂志。 2004; 24:5439–5444. doi:10.1523/JNEUROSCI.0974-04.2004。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Robinson SR,Bishop GM。Abeta作为生物絮凝剂:阿尔茨海默病淀粉样蛋白假说的含义。 神经生物老化。 2002; 23:1051–1072. doi:10.1016/S0197-4580(01)00342-6。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
主教GM,Robinson SR。淀粉样蛋白假说:让睡眠教条撒谎? 神经生物老化。 2002; 23:1101–1105. doi:10.1016/S0197-4580(02)00050-7。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Plant LD、Boyle JP、Smith IF、Peers C、Pearson HA。 淀粉样β肽的产生是中枢神经元存活的关键要求。 神经科学杂志。 2003; 23:5531–5535. doi:10.1523/JNEUROSCI.23-13-05531.2003。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Giulian D,Haverkamp LJ,Yu JH,Karshin W,Tom D,Li J.等人。阿尔茨海默病斑块中β-淀粉样蛋白的特定域引发人类小胶质细胞的神经元死亡。 神经科学杂志。 1996; 16:6021–6037. doi:10.1523/JNEUROSCI.16-19-06021.1996。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Giulian D、Haverkamp LJ、Yu J、Karshin W、Tom D、Li J.等人。β-淀粉样蛋白的HHQK结构域为阿尔茨海默病的免疫病理学提供了结构基础。 生物化学杂志。 1998; 273:29719–29726. doi:10.1074/jbc.273.45.29719。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Thomas T、Thomas G、McLendon C、Sutton T、Mullan M.β-淀粉样蛋白介导的血管活性和血管内皮损伤。 自然。 1996; 380:168–171. doi:10.1038/380168a0。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Paris D、Townsend K、Quadros A、Humphrey J、Sun J、Brem S.等。阿贝塔肽抑制血管生成。 血管生成。 2004; 7:75–85. doi:10.1023/B:AGEN.0000037335.17717.bf。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Roher AE、Lowenson JD、Clarke S、Woods AS、Cotter RJ、Gowing E.等人。β-淀粉样蛋白-(1-42)是脑血管淀粉样蛋白沉积的主要成分:对阿尔茨海默病病理学的影响。 美国国家科学院院刊1993; 90:10836–10840. doi:10.1073/pnas.90.22.10836。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Atwood CS、Bowen RL、Smith MA、Perry G.脑血管对密封剂、抗凝剂和重塑分子的要求,以维持血管完整性和血液供应。 脑研究脑研究修订版2003; 43:164–178. doi:10.1016/S0165-0173(03)00206-6。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Roskam S,Neff F,Schwarting R,Bacher M,Dodel R.APP转基因小鼠:主动和被动免疫治疗对认知任务的影响。 《神经科学生物学行为》2010年版; 34:487–499. doi:10.1016/j.neu-biorev.2009.10.006。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Loerch PM、Lu T、Dakin KA、Vann JM、Isaacs A、Geula C.等。衰老脑转录组的进化和突触调节。 公共科学图书馆一号。 2008; 3:e3329。 doi:10.1371/journal.pone.0003329。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Roher AE,Kokjohn TA。 AbetaPP转基因小鼠作为阿尔茨海默病淀粉样级联模型的评价。 当前医学化学免疫内分泌和代谢药物。 2003; 3:85–90。 [ 谷歌学者 ]
Kokjohn TA,Roher AE。淀粉样前体蛋白转基因小鼠模型和阿尔茨海默病:理解范式、局限性和贡献。 老年痴呆症。 2009; 5:340–347. doi:10.1016/j.jalz.2009.03.002。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Kokjohn TA,Roher AE。一项中断试验中AD患者的抗体反应、淀粉样β肽残留和AN-1792免疫的临床效果。 中枢神经系统神经紊乱药物靶点。 2009; 8:88–97. doi:10.2174/187152709787847315。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Nicoll JA、Wilkinson D、Holmes C、Steart P、Markham H、Weller RO。用淀粉样β肽免疫后人类阿尔茨海默病的神经病理学:病例报告。 《国家医学》,2003年; 9:448–452. doi:10.1038/nm840。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Ferrer I、Boada RM、Sanchez Guerra ML、Rey MJ、Costa-Jussa F.阿尔茨海默病淀粉样β免疫接种后脑炎的神经病理学和发病机制。 脑病理学。 2004; 14:11–20. doi:10.1111/j.1750-3639.2004.tb00493.x。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Masliah E、Hansen L、Adame A、Crews L、Bard F、Lee C.等人。阿贝塔疫苗在阿尔茨海默病无脑炎的情况下对斑块病理学的影响。 神经病学。 2005; 64:129–131. doi:10.1212/01.WNL.000148590.39911.DF。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Nicoll JA、Barton E、Boche D、Neal JW、Ferrer I、Thompson P.等人。阿贝塔42免疫后阿贝塔种的去除。 神经病理学实验神经学杂志。 2006; 65:1040–1048. doi:10.1097/01.jnen.0000240466.10758.ce。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Patton RL、Kalback WM、Esh CL、Kokjohn TA、Van Vickle GD、Luehrs DC。 等。AN-1792免疫性阿尔茨海默病患者的淀粉样β肽残留:生化分析。 《美国病理学杂志》。 2006; 169:1048–1063。 doi:10.2353/ajpath.2006.060269。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Holmes C、Boche D、Wilkinson D、Yadegarfar G、Hopkins V、Bayer A等。阿贝塔42免疫治疗阿尔茨海默病的长期效果:一项随机、安慰剂对照的I期试验的随访。 柳叶刀。 2008; 372:216–223. doi:10.1016/S0140-6736(08)61075-2。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Boche D、Zotova E、Weller RO、Love S、Neal JW、Pickering RM.等。Abeta免疫对人类阿尔茨海默病脑血管系统的影响。 大脑。 2008; 131:3299–3310. doi:10.1093/brain/own261。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Uro-Coste E、Russano de PG、Guilbeau-Frugier C、Sastre N、Ousset PJ、da Silva NA等。β淀粉样蛋白疫苗接种后的脑淀粉样血管病和微出血:病例报告和简要回顾。 临床神经病理学。 2010; 29:209–216. doi:10.5414/npp29209。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Orgogozo JM、Gilman S、Dartigues JF、Laurent B、Puel M、Kirby LC等。Abeta42免疫接种后AD患者亚急性脑膜炎。 神经病学。 2003; 61:46–54. doi:10.1212/01.wnl.0000073623.84147.a8。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Salloway S、Sperling R、Gilman S、Fox NC、Blennow K、Raskind M.等人。巴匹诺单抗治疗轻中度阿尔茨海默病的2期多剂量递增试验。 神经病学。 2009; 73:2061–2070. doi:10.1212/WNL.0b013e3181c67808。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Serrano-Pozo A、William CM、Ferrer I、Uro-Coste E、Delisle MB、Maurage CA等。人类抗淀粉样β主动免疫对神经突起形态和tau病理学的有益影响。 大脑。 2010; 133:1312–1327. doi:10.1093/brain/awq056。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Boche D、Donald J、Love S、Harris S、Neal JW、Holmes C.等人。阿贝塔42免疫治疗阿尔茨海默病后,神经元过程中聚集的Tau减少,但细胞体内的Tau没有减少。 神经病理学学报。 2010; 120:13–20. doi:10.1007/s00401-010-0705-y。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Beach TG、Sue LI、Walker DG、Roher AE、Lue L、Vedders L.等人。太阳健康研究所脑捐赠计划:描述和经验,1987-2007。 细胞组织库。 2008; 9:229–245. doi:10.1007/s10561-008-9067-2。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Mulugeta E、Molina-Holgado F、Elliott MS、Hortobagyi T、Perry R、Kalaria RN等。混合型和血管性痴呆患者额叶的炎症介质。 老年痴呆症认知障碍。 2008; 25:278–286. doi:10.1159/000118633。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Maarouf CL、Daugs ID、Spina S、Vidal R、Kokjohn TA、Patton RL等。与早老蛋白突变相关的痴呆患者的组织病理学和分子异质性。 摩尔神经变性剂。 2008; 3:20. doi:10.1186/1750-1326-3-20。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Esh C、Patton L、Kalback W、Kokjohn TA、Lopez J、Brune D.等人。PDAPP(Val717-->Phe)转基因小鼠中APP处理的改变产生了延长的Abeta肽。 生物化学。 2005; 44:13807–13819. doi:10.1021/bi051213+。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Dean RB、Dixon WJ。 针对少量观测的简化统计数据。 分析化学。 1951; 23:636–638. doi:10.1021/ac60052a025。 [ 内政部 ] [ 谷歌学者 ]
Roher AE、Lowenson JD、Clarke S、Wolkow C、Wang R、Cotter RJ.等。β-淀粉样蛋白核心蛋白的肽链结构改变可能是其在阿尔茨海默病中沉积和稳定的原因。 生物化学杂志。 1993; 268:3072–3083. [ 公共医学 ] [ 谷歌学者 ]
Kuo YM,Emmerling MR,Woods AS,Cotter RJ,Roher AE。神经炎斑块和血管淀粉样蛋白沉积物中Aβ3-丙谷氨酰肽的分离、化学表征和定量。 生物化学与生物物理研究委员会。 1997; 237:188–191. doi:10.1006/bbrc.1997.7083。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Kuo YM、Emmerling MR、Vigo-Pelfrey C、Kasunic TC、Kirkpatrick JB、Murdoch GH等。正常和阿尔茨海默病大脑中的水溶性阿贝塔(N-40,N-42)低聚物。 生物化学杂志。 1996; 271:4077–4081. doi:10.1074/jbc.271.8.4077。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Roher AE、Chaney MO、Kuo YM、Webster SD、Stine WB、Haverkamp LJ。 等。阿贝塔-(1-42)二聚体的形态和毒性,来源于阿尔茨海默病的神经炎和血管淀粉样沉积。 生物化学杂志。 1996; 271:20631–20635. doi:10.1074/jbc.271.34.20631。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Kuo YM、Webster S、Emmerling MR、De LN、Roher AE。不可逆二聚化/四聚化和翻译后修饰抑制阿尔茨海默病Aβ肽的蛋白水解降解。 Biochim生物物理学报。 1998; 1406:291–298. doi:10.1016/s0925-4439(98)00014-3。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Kerchner GA,Boxer AL.Bapineuzumab。 生物治疗专家。 2010; 10:1121–1130. doi:10.1517/14712598.2010.493872。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Scheinin NM、Aalto S、Koikkalainen J、Lotjonen J、Karrasch M、Kemppainen N等。阿尔茨海默病患者和对照组[11C]PIB摄取和脑容量的随访。 神经病学。 2009; 73:1186–1192. doi:10.1212/WNL.0b013e3181bacf1b。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Engler H、Forsberg A、Almkvist O、Blomquist G、Larsson E、Savitcheva I等。阿尔茨海默病患者淀粉样蛋白沉积的两年随访。 大脑。 2006; 129:2856–2866. doi:10.1093/brain/awl178。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Jack CR Jr、Lowe VJ、Weigand SD、Wiste HJ、Senjem ML、Knopman DS等。正常、轻度认知障碍和阿尔茨海默病的串行PIB和MRI:阿尔茨海默氏病病理事件序列的意义。 大脑。 2009; 132:1355–1365. doi:10.1093/brain/awp062。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
海宁格K。阿兹海默症的统一假说。 四、 事件的起因和顺序。 《神经科学评论》。 2000年,第213-328页。 [ 内政部 ] [ 公共医学 ]
Shankar GM、Li S、Mehta TH、Garcia-Munoz A、Shepardson NE、Smith I.等。直接从阿尔茨海默病患者大脑中分离的淀粉样β蛋白二聚体损害突触可塑性和记忆。 《国家医学》2008; 14:837–842. doi:10.1038/nm1782。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
廖磊,程德,王杰,Duong DM,Losik TG,Gearing M.等。激光捕获显微切割分离的死后淀粉样斑块的蛋白质组学特征。 生物化学杂志。 2004; 279:37061–37068. doi:10.1074/jbc。 M403672200。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Roher AE、Palmer KC、Yureucz EC、Ball MJ、Greenberg BD。从阿尔茨海默病脑组织中纯化的淀粉样斑块核心蛋白的形态和生化分析。 神经化学杂志。 1993; 61:1916–1926. doi:10.1111/j.1471-4159.1993。tb09834.x。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Wisniewski HM,Wegiel J.阿尔茨海默病的神经病理学。 《神经影像临床与美国》1995; 5:45–57. [ 公共医学 ] [ 谷歌学者 ]
神经病理学和神经炎症思想。 阿尔茨海默病杂志。 2009; 18:473–481. doi:10.3233/JAD-2009-1158。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Gowing E、Roher AE、Woods AS、Cotter RJ、Chaney M、Little SP等。Aβ17-42肽的化学表征,该肽是阿尔茨海默病弥漫性淀粉样沉积的组成部分。 生物化学杂志。 1994; 269:10987–10990. [ 公共医学 ] [ 谷歌学者 ]
Wei W、Norton DD、Wang X、Kusiak JW。 阿尔茨海默病中的Abeta 17-42激活JNK和caspase-8,导致神经元凋亡。 大脑。 2002; 125:2036–2043. doi:10.1093/brain/awf205。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Racke MM、Boone LI、Hepburn DL、Parsadainian M、Bryan MT、Ness DK等。免疫治疗对淀粉样前体蛋白转基因小鼠脑淀粉样血管病相关微出血的加重依赖于对淀粉样β沉积形式的抗体识别。 神经科学杂志。 2005; 25:629–636. doi:10.1523/JNEUROSCI.4337-04.2005。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Wilcock DM,Colton CA。转基因小鼠的免疫治疗、血管病理学和微出血。 中枢神经系统神经紊乱药物靶点。 2009; 8:50–64. doi:10.2174/187152709787601858。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Burbach GJ、Vlachos A、Ghebremedhin E、Del Turco D、Coomaraswamy J、Staufenbiel M.等。APP23转基因小鼠被动抗Abeta免疫治疗后的血管超微结构和随后的脑出血。 神经生物老化。 2007; 28:202–212. doi:10.1016/j.neurobiolaging.2005.12.003。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Lue LF,Walker DG。 阿尔茨海默病免疫治疗机制建模:人类死后小胶质细胞与抗体-淀粉样β肽的相互作用。 神经科学研究杂志,2002年; 70:599–610. doi:10.1002/jnr.10422。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]
Yamamoto M、Kiyota T、Walsh SM、Liu J、Kipnis J、Ikezu T。细胞因子介导的人类单核吞噬细胞对纤维淀粉样β肽降解的抑制。 免疫学杂志。 2008; 181:3877–3886. doi:10.4049/jimmunol.181.6.3877。 [ 内政部 ] [ PMC免费文章 ] [ 公共医学 ] [ 谷歌学者 ]
Blessed G,Tomlinson BE,Roth M.老年人大脑灰质中痴呆症定量测量值与老年性变化之间的关系。 Br J精神病学。 1968; 114:797–811。 doi:10.1192/bjp.114.512.797。 [ 内政部 ] [ 公共医学 ] [ 谷歌学者 ]