摘要
选择性剪接是一种普遍存在的转录后过程,它不仅对正常细胞功能很重要,而且还与人类疾病有关。新开发的第二代测序技术提供了高通量数据(RNA-seq数据)来研究不同类型细胞中的选择性剪接事件。在这里,我们提出了一种计算方法SpliceMap,用于从RNA-seq数据中检测剪接连接。该方法不依赖于任何现有的基因结构注释,能够发现具有高灵敏度和特异性的新剪接连接。它可以处理长读取(50–100 nt),并可以利用成对读取信息来提高映射精度。输出中包含多个参数,以指示预测结的可靠性,并帮助过滤错误预测。我们应用拼接图分析了2300万对来自人脑组织的50-nt读数。结果表明,在测序的这个深度,RNA-seq可以支持对剪接连接的可靠检测,但那些存在于非常低水平的剪接连接除外。与当前方法相比,拼接图可以在不牺牲特异性的情况下实现12%的高灵敏度。
简介
RNA剪接是一个重要的转录后步骤,其中前信使核糖核酸的一个或多个片段被剪接出来,其余片段(外显子)被连接以形成成熟的信使核糖核酸产物。通过选择性剪接,可以从同一遗传位点产生不同的转录物(亚型)。这个过程发生在90%以上的多基因人类中(1,2)并大大增加了转录组中可能转录物的多样性。已发现异常RNA剪接与许多人类疾病有关(三,4). 因此,识别和量化剪接事件的技术对生物学和医学很重要。
研究剪接转录物结构和丰度的最流行方法是通过对表达序列标签(EST)进行测序(5). 传统上,由于桑格法(EST项目中使用的主要测序技术)的低吞吐量,此类研究成本高昂且效率低下。然而,随着第二代测序技术(SGS)的出现,现在以高通量和经济高效的方式对转录组进行深入和全面的测序是可行的(6–8),使检测罕见的选择性剪接事件成为可能。在这种RNA-seq项目中,从研究中的转录物中随机读取数千万或数亿个短序列(30-100 nt)。因此,分析的第一步是将每个短读数映射到参考基因组,以确定可能导致这种读数的遗传位点。对于完全在外显子区域内采样的读取,此映射任务可以由任何现有的短读映射程序处理,例如ELAND(Cox,未发布的软件)和SeqMap(9).
然而,我们对新亚型发现最感兴趣的阅读是跨越外显子-外显子连接的阅读。这些“连接读取”不能直接映射到基因组。一种方法是将读取结果映射到当前注释外显子库中已知的转录序列。由于外显子库是不完整的,这种方法无法找到涉及新剪接事件的连接(10). 在另一种方法中,用于最近开发的TopHat(11)程序中,可在参考基因组上映射的读取被分组为不同的簇,以便每个簇中的读取通过重叠区域链接在一起。然后,每个簇定义一个假定的外显子区域。随后,可以根据这些假定的外显子定义来搜索外显子-外显子连接。
聚类是在第一个RNA-seq实验中发现新连接的自然方法(1,6,10,12–17)因为在SGS开发的早期阶段生成的数据大多是非常短的读数(25-36nt),不适合直接读取从头开始外显子-外显子连接的检测。然而,该技术正在迅速改进,目前一些SGS仪器(如Illumina Genome Analyzer)的可用读取长度通常在50–100 nt之间。增加的读取长度为直接绘制外显子-外显子连接图提供了可能,而无需参考任何假定或注释外显子。在这里,我们报告了一种新的算法,该算法基于使用半衰期映射的思想,以确定连接的近似位置。此外,该方法可用于合并配对测序数据中包含的额外信息,以获得比单端测序更高的特异性。该方法是在一个名为SpliceMap的免费Python程序中实现的(http://biogibs.stanford.edu/~kinfai/拼接图/).
材料和方法
拼接图仅利用参考基因组序列来独立于现有外显子注释来找到连接。如果测序足够深入,就有可能探索所有外显子剪接事件,包括已知和新的外显子。核心概念是在绘制完整连接之前,首先在剪接事件中涉及的两个外显子之一上确定连接边界。跨越连接的读取必须在参考基因组中具有不短于其一半长度的匹配。这样的匹配提供了一个种子,可以用来识别一个小的基因组区域,以搜索相应的连接。SpliceMap有四个主要步骤:半读映射、种子选择、连接搜索和配对过滤(图1). 当数据未配对结束时,不应用最后一步。对于大于50 nt的读取,我们从中提取几个重叠的50 nt读取,然后应用标准方法。例如,我们将100-nt读取拆分为三个段(1-50、26-75和51-100)。在长读取数据的后处理步骤中添加了一个额外的过滤器,以使用全长信息检查结果。通过这种方式,我们可以从单个长读取中找到多个连接。
图1。
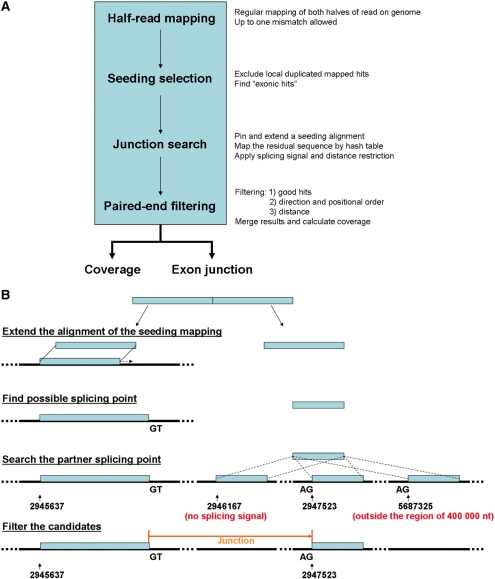
标准拼接图的工作流程和基于半读图的交叉搜索轮廓:(一)拼接图由四个步骤组成:半读映射、种子选择、连接搜索和配对过滤。拼接图输出覆盖图和检测到的连接。(b条)每个半读都与基因组对齐,并进行扩展以获得部分对齐。读取的剩余部分(如果至少为10 nt)将用于搜索邻域(40万nt)内的匹配项。GT–AG拼接信号也用于筛选匹配项。
半读映射
利用最新的第二代测序器模型提供的合理长的读取(50 nt),半长(25 nt)可以以较高的概率与参考基因组序列进行可靠的比对。在这一步中,SpliceMap通过任何当前可用的短读映射工具(如SeqMap)将读取的两部分映射到参考基因组(9)和ELAND。根据数据质量和读取长度,可以相应地选择半读映射允许的最大不匹配。映射后,将逐条染色体执行以下步骤。
播种选择
我们使用半读的映射命中来缩小结的搜索区域。在接下来的步骤中,这些命中将逐个基础地扩大。因此,我们将半读映射命中称为“种子”。将检查上述步骤中映射的命中数以进行种子选择。虽然唯一映射的点击数与接合搜索的种子相比更可靠,但不应简单地排除所有多重映射读取(即映射到多个位置的读取),因为这样做将大大减少检测其他地方同源序列的接合的机会,例如同源基因或假基因中的接合。SpliceMap没有拒绝所有的多重映射点击,而是只排除那些与同一半读中的另一个点击量相差40万nt以内的点击。因为如果两个区域在40万nt的距离内是相同的,那么在这两个区域之间往往会形成错误的剪接预测,而这两个区与读数完全匹配。
交叉路口搜索
对于每一个确定的种子,参考基因组上的比对都会逐个碱基扩展以找到剪接点(图1). SpliceMap随后尝试在用户指定的距离内(在我们的示例中设置为40万nt)找到伙伴拼接点,该拼接点与原始读取的相应剩余序列完美匹配。当完整读数为50 nt时,拼接点候选必须满足两个标准:第一,对齐延伸不能超过40 nt,剩余长度必须至少为10 nt;其次,剪接点必须紧邻供体和受体位点的规范二核苷酸剪接信号GT和AG,因为它们出现在98%的已知剪接位点中(18). 残差序列的映射是通过在预先计算的染色体宽度散列表中搜索10-nt种子,然后扩展以完成完全对齐来实现的。为了减少假阳性连接,如果搜索产生满足上述标准的剩余序列的多个匹配,则会丢弃结果。
成对end滤波
当配对读取可用时,此步骤中使用配对信息以提高连接检测的特异性。首先,在前面的步骤中,三种类型的点击被确定为“好点击”,即外显点击、扩展点击和连接点击。如果完全读取的两部分被映射到正好相差读取长度一半的位置,则会发生外显子命中。另一方面,如果半读命中可以最大限度地扩展到一个适当长但比完整读取长度短的对齐长度,则将其视为扩展命中。最后,如上所述确定连接命中。为了符合可靠点击的条件,从成对读取的两次读取中生成的点击必须满足以下条件(i)两次点击都是“良好点击”;(ii)其距离不超过400000 nt;(iii)两次点击的参考基因组序列的定位方向和位置顺序与实验设计的顺序一致(图2).
图2。

成对输入的方向和位置顺序读取(R1–R2)。如果测序样品与原始副本相同,则读取的R1应可在5′端正向映射,R2应可在3′端反向映射。如果测序样品是互补副本,则读取的R1应可在3′端反向映射,R1应在5′端正向映射。
可靠性评估参数和可选滤波
拼接图提供了几个参数,以便于评估检测到的连接的可靠性和丰富性。对于每个连接,SpliceMap计算其nR(支持此连接的读取数)、nNR(非冗余支持读取数),nUM(唯一映射支持读取数,nUP和nDOWN(分别在上游或下游相邻区域中的映射读取数)(图3). 我们在Illumina提供给我们的23 412 226对50-nt配对读取的RNA-seq数据集上检查了这些参数。这些参数的不同要求导致不同的可选参数滤波。
图3。
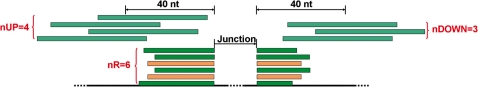
拼接图中评估连接的参数示意图。深绿色读取是唯一映射的支持读取(nUM=4),而小麦读取是乘法映射的支持读。因此,该交叉点的nR为6。但一些支持读取是多余的,因此nNR=4。在40 nt的上游和下游相邻区域分别有四个和三个唯一映射读取(灰绿色),因此nUP=4和nDOWN=3。
我们发现,对于RefSeq注释为只有一个亚型的基因上靠近3′端的剪接连接,支持读数(nR)与总表达显著相关(对2=0.7173),以RPKM测量的基因(每百万可映射读数中每kb转录本的读数)(10). 这表明,当交叉点可能发生选择性剪接时,我们可以使用nR作为其使用的指标。表1显示了在任何可选参数滤波之前预测结之间的nR分布。虽然总的来说只有29%的预测结具有nR=1,但在新结中这一百分比增加到74%。这与当前注释中未包含的连接可能相对罕见的概念一致。
表1。
根据成对输入数据(无任何可选参数滤波)用SpliceMap预测所有结之间和新结之间的nR分布
| 无风险 |
所有交叉口 |
新型结一
|
| 1 |
49 888 (28.75%) |
26 744 (74.10%) |
| 2–5 |
49 462 (28.51%) |
7942 (22.00%) |
| 6–20 |
44 577 (25.69%) |
1222 (3.39%) |
| 21 ∼ 50 |
19 074 (10.99%) |
147 (0.41%) |
| 51 ∼ 200 |
10 478 (6.04%) |
34 (0.09%) |
| 201 ∼ 1000 |
1787 (1.03%) |
2 (0.01%) |
| 1000+ |
135 (0.08%) |
0 (0%) |
连接的nR受待测序库中RNA片段表示或扩增的随机性影响。如果RNA片段很容易被文库制备协议捕获(本例中为随机六聚体启动),然后高度扩增,则很可能产生相同序列的多次读取。我们称这些为“冗余”读取。对于低丰度异构体中的连接,如果两个或多个支持读序列相同,则它们可能是多余的。在这种情况下,即使nR>1,我们仍然可能没有可靠的预测。通过要求连接由多个“非冗余”读取支持(nNR>1),我们可以大大提高预测的特异性。我们可以看到,一旦我们要求nNR>1(114 990/121 718=94%),EST验证率就非常高。当我们仅限于新型结时,nNR>1的预测的EST验证仍然很高(4494/7757=58%)(表2). 由于EST数据对罕见亚型中新连接的覆盖可能不完整,nNR>1预测的特异性应大大高于58%。我们将在讨论实验验证结果时进一步研究这个问题。
表2。
|
拼接贴图
|
| 可选过滤器#
|
– |
号码 |
下载(UP/nDOWN) |
无核反应堆 |
nUM+nUP/nDOWN |
| 总交叉口 |
175 401 |
168 807 |
162 060 |
121 718 |
151 317 |
| 新型结 |
36 091 |
32 060 |
27 497 |
7757 |
23 020 |
| 与EST验证的连接 |
145 517 |
142 610 |
139 880 |
114 990 |
133 010 |
| 新型EST连接 |
12 053 |
11 549 |
10 562 |
4494 |
9493 |
| EST验证率 |
82.96% |
84.48% |
86.31% |
94.47% |
87.90% |
| EST验证率(新) |
33.40% |
36.02% |
38.41% |
57.93% |
41.24% |
尽管要求nNR>1可以有效地确保结预测的高度特异性,但它将预测的结数量减少了约31%。在新的预测中,潜在的灵敏度损失甚至更大,即从36 091到7757。这促使我们设计可选滤波器,以区分nNR=1的结之间的可靠预测和不可靠预测。我们要求连接必须由至少一个唯一映射读取(nUM>0)支持,而不是通过nR或nNR过滤连接预测。这一要求排除了3.8%的连接,但将总特异性从82.96%提高到84.48%(表2).
另一个可选过滤器是在假定接头的每一侧,在拼接位置附近映射足够数量的读取。要做到这一点,请记住,nUP是一个小区域内的读取数(外显子或连接读取数)(例如,邻域大小为K(K)nt)转录本中感兴趣的剪接位点的上游,nDOWN是相应的下游相邻读。因此,我们的过滤器要求每个nUP或nDOWN至少为1。表3显示了检测到的连接数和邻域大小的EST验证率K(K)=40、80和160。我们可以看到,这种过滤排除了一些连接,但提高了特异性K(K)减少。通过调整K(K),我们可以在连接预测中实现灵敏度和特异性的适当平衡。什么时候?K(K)=40,排除7.6%的连接,但总特异性从82.96%增加到86.91%(表3). 下面比较了各种可选参数滤波器的增益和损耗表2.
表3。
在标准配对端拼接映射之后,用于nUP/nDOWN过滤的各种邻域大小的检测到的结的数量和EST验证率
|
K(K)= 40 |
K(K)= 80 |
K(K)= 160 |
| 总交叉口 |
156 015 |
162 060 |
165 452 |
| 新型结 |
25 441 |
27 497 |
29 342 |
| 与EST验证的连接 |
135 586 |
139 880 |
141 758 |
| 新型EST连接 |
9901 |
10 562 |
11 062 |
| EST验证率 |
86.91% |
86.31% |
85.68% |
| EST验证率(新) |
38.92% |
38.41% |
37.70% |
结果
特异性
我们在Illumina提供给我们的23 412 226对50-nt配对读取的RNA-seq数据集上测试了拼接地图和TopHat。用寡核苷酸-dT磁珠从人类脑组织总RNA中纯化mRNA。用随机引物引物合成cDNA。按照制造商的建议,使用Illumina基因组分析仪对ds–cDNA进行测序。该数据在GEO数据库中公开,并带有登录号GSE19166标准在本测试中,拼接图要求读取内容覆盖接合处每侧至少10 nt。基于25-nt半衰期的种子映射中允许存在一个不匹配,但剩余序列的映射需要完全匹配。种子映射上的错配允许检测侧翼外显子序列中的SNP连接,也允许测序错误。
在这里,我们报告了通过配对拼接图和nUP/nDOWN过滤发现的结果(K(K)=40)和nUM过滤。SpliceMap共发现151 317个外显子连接,包括23020个新连接,这在RefSeq中没有报道(19)、合奏(20)和KnownGene(21). 为了评估拼接图的特异性,检测到的连接与GenBank中的人类EST对齐(22). 因为EST是200-600 nt的单次读取,所以它们通常足够长,能够高可靠性地识别独特的转录片段。因此,如果可以在EST序列中找到我们检测到的连接的匹配从头开始剪接发现算法,那么这可以看作是对连接存在性的独立实验验证。作为比较,我们还展示了TopHat 1.0.12的相应结果(11)(截至2009年10月28日的最新版本)(表4和图4).
表4。
在nUM和nUP/nDOWN之后,TopHat和SpliceMap的结果统计(K(K)=40)过滤
|
拼接贴图 |
顶帽 |
| 总交叉口 |
151 317 |
133 722 |
| 新型结一
|
23 020 |
19 777 |
| 与EST验证的连接b条
|
133 010 (87.90%) |
117 113 (87.58%) |
| nNR>1的连接(多个非冗余读取) |
119 298 |
– |
| 与EST的新型连接 |
9493 (41.24%) |
7273 (36.78%) |
| nNR>1的新型结 |
7187 |
– |
| 与EST和nNR>1的交界处 |
112 962 |
– |
| EST和nNR>1的新型结 |
4242 |
– |
图4。
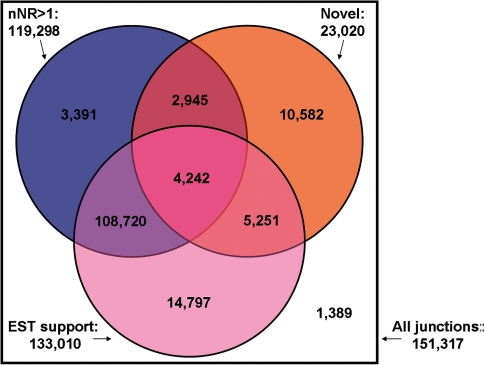
拼接图预测的连接的冗余分布、新颖性和EST证据的维恩图。只有1389个已知交叉点具有单个非冗余读取,并且没有EST证据支持。
我们发现拼接图检测到的连接中87.9%(133010)得到EST证据的支持。如果我们仅限于SpliceMap预测的23020个新连接,则百分比会降低(41.2%)(9493个连接),但仍表示EST支持的合理程度。我们注意到,EST序列不是转录组的全面表示,EST支持的连接百分比只是对特异性的保守估计。我们可以看到,与TopHat相比,SpliceMap具有类似的EST验证率,但检测到更多的连接(151 317对133 722)。
敏感
表5给出了拼接图检测RefSeq中具有单个亚型的12755个基因连接的灵敏度。根据相应基因的表达水平(以RKPM为单位)对连接进行分类,并计算每个分类的检测率。可以看出,SpliceMap对高表达基因(RKPM>20)具有极高的灵敏度(>95%)。对于中等表达(5<RKPM<20)的基因,敏感性仍然很高(90%),而对于相对低表达(1<RKPM<5)的基因来说,敏感性仍然很大(40–67%)。然而,对于表达非常低(RKPM<1)的基因,敏感性降至7%以下。我们的结果表明,要分析这种罕见的转录本,需要比2300万对读更深入的测序。最后,表5此外,在所有基因表达水平上,与当前方法(TopHat)的代表相比,拼接图可以检测更多RefSeq注释连接。
表5。
|
拼接图(%) |
顶帽(%) |
| 0<RPKM≤1(2993) |
6.76 |
5.54 |
| 1<RPKM≤2(1199) |
40.86 |
27.74 |
| 2<RPKM≤5(2049) |
67.23 |
52.24 |
| 5<RPKM≤20(3245) |
89.55 |
80.95 |
| 20<RPKM≤50(1340) |
95.55 |
91.10 |
| 50<RPKM≤100(522) |
97.18 |
93.87 |
| RPKM>100(408) |
95.66 |
88.58 |
我们还检查了具有单个RefSeq亚型的基因的连接恢复程度。对于每个基因,我们通过计算检测到的连接百分比来衡量其连接发现程度。结果显示在表6我们可以看到,拼接图检测到的更多基因在连接发现中具有更高的完整度(80–100%)。特别是,这种完整性在下游分析中可能很重要,例如异构体重建和丰度估计。
表6。
|
拼接贴图 |
顶帽 |
| 检测到的基因数量一
|
8939 |
8777 |
| 1 ≤P(P)b条< 50 |
1076 |
1729 |
| 51 ≤P(P)< 80 |
1812 |
2319 |
| 81 ≤P(P)<100 |
1600 |
1347 |
|
P(P)= 100 |
4451 |
3382 |
PCR验证
除了EST的证据外,我们的新连接还通过PCR实验进行了验证。随机选择了20个预测,包括18个新的外显子跳跃事件和两个新外显子检测,所有预测都没有任何当前已知的EST或人类mRNA证据。18个新颖的跳过事件来自五个具有不同非冗余读取数量的容器:1、2、3-5、6-10和>10。从每个箱子中随机选择三到四个预测。PCR验证表明,17个(85%)得到验证。在三个错误预测中,有两个出现在单个非冗余读取的容器中。因此,在nNR>1的新预测中,PCR验证率为13/14=92.86%。这明显更高(使用Z轴测试P(P)-值为0.004),而EST验证率为58%,我们认为这低估了特异性,因为EST对罕见亚型的覆盖不完整。
与ERANGE的比较
查看SpliceMap与使用参考注释的简单方法的比较;我们还运行了一个依赖于注释的工具ERANGE(10)在同一数据集上。ERANGE识别出160 899个连接点,其中127 043个连接点也被SpliceMap检测到。ERANGE发现的所有结果都不新颖,但包括一些拼接图未检测到的连接,因为ERANGE放宽了对次要侧翼序列长度的要求。ERANGE在每个侧翼序列上需要至少4nt,而剪接映射需要至少10nt。更严格的要求保证了拼接图预测新型结的合理可靠性。新的连接发现是拼接地图的主要功能,因此不能被独立于注释的ERANGE所取代。在SpliceMap发现的151 317个连接中,有24 274个没有被ERANGE报道,其中23020个是新的。
与BLAT的比较
我们还将拼接图与BLAT进行了比较,BLAT是一种常用于对齐EST序列的工具。为了进行公平的比较,我们优化了BLAT中的参数设置,并通过要求存在典型拼接信号来过滤结果。我们发现,当BLAT以最非精简的参数运行且无过滤时,其灵敏度为SpliceMap的~55%。然而,在这种非特异性环境中,BLAT检测到的连接大多为假阳性(特异性仅为3%)。通过更严格的参数和添加后对齐过滤步骤,我们可以大大提高特异性,但代价是进一步降低灵敏度。在最佳设置下,BLAT的特异性水平与SpliceMap相似,但仍略低,敏感性低得多(低70%)。Illumina Brain数据集上两种方法之间的比较细节见表7.
表7。
|
BLAT(爆炸) |
拼接贴图 |
| 最大内含子大小(nt) |
750 000 |
750 000 |
400 000 |
400 000 |
400 000 |
| 瓷砖尺寸(nt) |
8–12 |
8–12 |
12 |
12 |
不适用 |
| Minscore公司 |
30 |
30 |
50 |
50 |
不适用 |
| 不匹配 |
1 |
1 |
0 |
0 |
1 |
| 接头数量 |
– |
1 |
1 |
1 |
1 |
| 侧翼序列的最小长度。 |
– |
10 |
10 |
10 |
10 |
| 标准拼接信号 |
– |
– |
– |
必修的 |
必修的 |
| 总交叉口 |
2 404 632 |
345 195 |
209 673 |
51 325 |
160 076 |
| 有效交叉点 |
75 147 |
61 671 |
45 714 |
41 173 |
137 965 |
| 特异性 |
3.12% |
17.87% |
21.80% |
80.22% |
86.19% |
BLAT是一种优化良好的比对工具,但它并不是专为从短阅读中检测外显子连接而设计的。SpliceMap和BLAT都是基于使用读取段(种子)的快速映射来缩小搜索区域,以便更详细地对齐读取。作为一种针对短RNA-seq数据的剪接检测工具,SpliceMap使用25-nt种子,而BLAT通常使用大约8-12-nt种子。SpliceMap通过使用短读对齐工具(如ELAND和SeqMap)生成种子,而BLAT使用哈希表。短播种允许BLAT发现跨越小外显子的剪接,但会导致许多错误的映射。如上所述,这些错误预测可以在基于典型剪接信号和配对信息的后处理步骤中检测和消除。然而,即使BLAT以最宽容的参数设置运行,后处理过滤器也无法弥补BLAT中灵敏度的损失。BLAT有自己的特殊后处理步骤,这些步骤不是专门为外显子连接检测设计的。当读数较短时,这些步骤可能会删除许多正确的拼接。尽管我们预计BLAT的性能可能会随着读取长度的增加而提高,但对于当前的RNA-seq数据,它无法取代定制设计的连接检测工具,如剪接映射。
运行时间
SpliceMap的更好性能是以更长的运行时间为代价的,它需要66个CPU小时来处理所有2300万次读取。相比之下,TopHat花了12个CPU小时来处理相同的数据集。然而,值得指出的是,这可能是因为SpliceMap是用Python编写的,而TopHat是用C++编写的。此外,在我们的实验中,我们使用了一个16核服务器,从而大大减少了总体运行时间。SpiceMap的Python部分已经用C++重写,速度提高了约4倍。
讨论
使用具有长读数(50 nt或更长)的配对末端测序有望大大提高我们表征转录物异构体的能力,但只有在有合适的计算方法来分析数据的情况下,这一承诺才能实现。在本文中,我们报告了50-nt读取可以支持直接从头开始剪接连接的检测不需要首先进行聚类读取来识别假定的外显子,并且这种方法可以在连接检测中实现比当前领先的RNA-seq分析方法更高的灵敏度。我们的结果通过对一个新数据集的系统分析得到了证实,该数据集包含2300万对来自人类脑组织的50-nt配对读取数据。使用EST序列作为验证,我们发现如果预测得到至少两个非冗余读取(nNR>1)的支持,我们的方法SpliceMap可以以非常高的验证率(95%)预测连接。然而,如果我们使用适当的附加过滤器来消除不可靠的预测,即使只有单个非冗余读取(nNR=1)支持的预测也可能具有很高的特异性。包括通过这些过滤器的nNR=1预测大大提高了检测灵敏度,并且仍然保持良好的验证率(87.9%)。此外,EST验证率可能会低估新预测的特异性,这一点通过我们使用基于PCR的方法对新预测子集进行的实验测试得到了证实。
RNA-seq结合点检测的灵敏度问题是一个重要问题,目前的文献尚未彻底研究。构建这个问题的一种方法是,对于给定的排序深度,C是什么50它被定义为每个单元拷贝数的下限,以便给定的连接有50%的机会被检测到。基于一个RKPM对应于本研究中所用协议的每个细胞约0.3个拷贝的假设(10),我们可以根据表5对于深度为2300万配对50-nt读取的RNA-seq,C50大约是每个单元格一个副本。
我们发现成对阅读信息有助于减少错误发现。对于用于生成数据的Illumina协议,mRNA中两个成对-end读取之间的距离约为200 nt,有时两个读取之间可能存在内含子。由于绝大多数内含子的大小都小于40万nt,拼接图将从所有后续计算中筛选出任何相隔40万nt以上的成对点击。图5显示了一个通过使用配对过滤消除错误预测的示例。与仅使用单个读取(即将同一数据集视为4600万个未配对50-nt读取)相比,使用配对过滤将所有预测的EST验证率从86.19%提高到87.9%,将新预测的验证率从38.53%提高到41.24%,在不影响检测注释连接的灵敏度的情况下。尽管单端拼接地图的性能仍优于TopHat,但我们建议研究人员尽可能使用配对测序。由于EST验证率低估了新预测的真实特异性,因此错误预测的减少应该比上述数字所显示的更大。
图5。
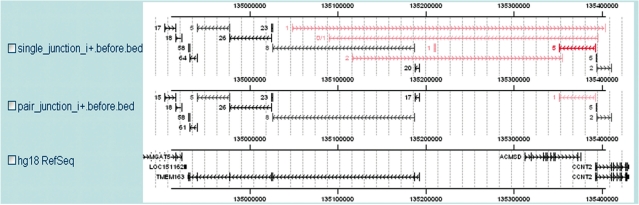
按成对输入信息过滤。前两个轨迹分别是在进行nUP、nDOWN和nUM过滤之前,单端拼接贴图和配对拼接贴图的结果。检测到的已知连接是黑色的,而新的连接是红色的。单读分析预测了几个非常长的跨基因连接。这些都是假阳性结果,配对信息有助于消除它们。
基金
美国国立卫生研究院(1R01HG004634 to W.H.W.);Edward Mallinckrodt Jr.基金会向Y.X.提供的初级教员补助金,用于开放存取费用:1R01HG004634。
利益冲突声明。未声明。
作者贡献
KFA、HJ和WHW构思了这项研究,设计了算法并进行了数据分析。KFA实施了算法并起草了手稿。HJ和WHW修改了手稿。L.L和XY。进行了实验验证。
鸣谢
Illumina Inc.的Shujun Luo和Gary Schroth提供了RNA测序数据。加里·施罗斯也对手稿提出了有益的评论。
参考文献
-
1Wang ET、Sandberg R、Luo S、Khrebtukova I、Zhang L、Mayr C、Kingsmore SF、Schroth GP、Burge CB。人类组织转录体中的替代亚型调控。自然。2008;462:470–476. doi:10.1038/nature07509。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
2Matlin AJ、Clark F、Smith CW。理解选择性剪接:走向细胞代码。自然反相摩尔电池。生物学2005;6:386–398. doi:10.1038/nrm1645。[内政部] [公共医学] [谷歌学者]
-
三。Nagao K、Togawa N、Fujii K、Uchikawa H、Kohno Y、Yamada M、Miyashita T。用外显子连接微阵列检测PTCH基因的组织特异性选择性剪接和疾病相关异常剪接。嗯,分子遗传学。2005;14:3379–3388. doi:10.1093/hmg/ddi369。[内政部] [公共医学] [谷歌学者]
-
4Wang H、Hubbell E、Hu JS、Mei G、Cline M、Lu G、Clark T、Siani-Rose MA、Ares M、Kulp DC等。使用微阵列平台进行基于基因结构的剪接变体反褶积。生物信息学。2003;19:315–322. doi:10.1093/bioinformatics/btg1044。[内政部] [公共医学] [谷歌学者]
-
5Adams MD、Soares MB、Kerlavage AR、Fields C、Venter JC。从定向克隆的人类婴儿大脑cDNA文库中快速测序(表达序列标签)。自然遗传学。1993;4:373–380. doi:10.1038/ng0893-373。[内政部] [公共医学] [谷歌学者]
-
6Marioni JC、Mason CE、Mane SM、Stephens M、Gilad Y.RNA-seq:技术再现性评估和与基因表达阵列的比较。基因组研究2008;18:1509–1517. doi:10.1101/gr.079558.108。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
7Shendure J.微阵列终结的开始?自然方法。2008;5:585–587. doi:10.1038/nmeth0708-585。[内政部] [公共医学] [谷歌学者]
-
8Wang Z,Gerstein M,Snyder M.RNA-Seq:转录组学的革命性工具。Nat.Rev.基因。2009;10:57–63. doi:10.1038/nrg2484。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
9姜浩、王浩。SeqMap:将大量寡核苷酸映射到基因组中。生物信息学。2008;24:2395–2396. doi:10.1093/bioinformatics/btn429。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
10Mortazavi A、Williams BA、McCue K、Schaeffer L、Wold B.通过RNA-Seq对哺乳动物转录体进行定位和量化。自然方法。2008;5:621–628. doi:10.1038/nmeth.1226。[内政部] [公共医学] [谷歌学者]
-
11Trapnell C,Pachter L,Salzberg SL。TopHat:发现与RNA序列的剪接连接。生物信息学。2009;25:1105–1111. doi:10.1093/bioinformatics/btp120。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
12Nagalakshmi U,Wang Z,Waern K,Shou C,Raha D,Gerstein M,Snyder M。通过RNA测序确定的酵母基因组转录图谱。科学。2008;320:1344–1349. doi:10.1126/science.1158441。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
13Wilhelm BT、Marguerat S、Watt S、Schubert F、Wood V、Goodhead I、Penkett CJ、Rogers J、Bähler J.以单核苷酸分辨率测量的真核转录组的动态库。自然。2008;453:1239–1243. doi:10.1038/nature07002。[内政部] [公共医学] [谷歌学者]
-
14Lister R、O'Malley RC、Tonti-Filippini J、Gregory BD、Berry CC、Millar AH、Ecker JR。拟南芥表观基因组高度集成的单碱基分辨率图谱。单元格。2008;133:523–536. doi:10.1016/j.cell.2008.03.029。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
15Cloonan N、Forrest AR、Kolle G、Gardiner BB、Faulkner GJ、Brown MK、Taylor DF、Steptoe AL、Wani S、Bethel G等。通过大规模mRNA测序进行干细胞转录组分析。自然方法。2008;5:613–619. doi:10.1038/nmeth.1223。[内政部] [公共医学] [谷歌学者]
-
16Pan Q,Shai O,Lee LJ,Frey BJ,Blencowe BJ。通过高通量测序深入研究人类转录组中的选择性剪接复杂性。自然遗传学。2008;40:1413–1415. doi:10.1038/ng.259。[内政部] [公共医学] [谷歌学者]
-
17Sultan M、Schulz MH、Richard H、Magen A、Klingenhoff A、Scherf M、Seifert M、Borodina T、Soldatov A、Parkhomchuk D等。通过人类转录组的深度测序对基因活性和选择性剪接的全球观点。科学。2008;321:956–960. doi:10.1126/science.1160342。[内政部] [公共医学] [谷歌学者]
-
18Burset M、Seledtsov IA、Solovyev VV。哺乳动物基因组中典型和非典型剪接位点的分析。核酸研究2000;28:4364–4375. doi:10.1093/nar/28.21.4364。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
19Pruitt KD、Tatusova T、Maglott DR.NCBI参考序列(RefSeq):基因组、转录物和蛋白质的精选非冗余序列数据库。核酸研究2005;33:501–504. doi:10.1093/nar/gkl842。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
20Curwen V、Eyras E、Andrews TD、Clarke L、Mongin E、Searle SM、Clamp M。Ensembl自动基因注释系统。基因组研究2004;14:942–950. doi:10.1101/gr.1858004。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
21Hsu F、Kent WJ、Clawson H、Kuhn RM、Diekhans M、Haussler D。UCSC已知基因。生物信息学。2006;22:1036–1046. doi:10.1093/bioinformatics/btl048。[内政部] [公共医学] [谷歌学者]
-
22Boguski MS、Lowe TM、Tolstoshev CM.dbEST–“表达序列标签”数据库。自然遗传学。1993;4:332–333. doi:10.1038/ng0893-332。[内政部] [公共医学] [谷歌学者]


