蛋白激酶C的关键步骤之一(PKC)2激活是它从细胞质转移到细胞膜。对于传统(α,βI,βII和γ)和新的(δ,ε,η,和θ)PKC,这种易位是由与亲脂性第二信使锡-1,2-二酰基甘油(DAG),生成从磷脂酰肌醇4,5-二磷酸活化受体偶联磷脂酶C或间接来自磷脂酰胆碱磷脂酶D(1). 一双PKCs调控结构域中的锌指结构“C1”域,负责识别DAG信号。DAG-C1域-膜相互作用耦合到构象PKC的变化,都导致假底物结构域从激活酶并触发向薄膜(2). 通过调节获得底物,PKC易位补充内在酶PKC的特异性,以确定其底物轮廓。
C1结构域是一个高度保守的富含半胱氨酸的基序(~50个氨基酸酸),在PKC中首次确定为DAG或佛波酯(三). 它具有球状结构,一端有亲水性结合裂缝被疏水残基包围。DAG或佛波酯与C1的结合结构域覆盖亲水裂缝并形成连续的疏水表面有利于C1结构域相互作用或渗透到膜中(4). 除了小说和经典PKC,其他六个蛋白质家族也已被鉴定,其中一些成员拥有DAG/phorbol酯类反应C1结构域。这些是蛋白激酶D吗(5),奇马林(6),munc-13(7)RasGRP(鸟苷Ras和Rap1的核苷酸交换因子(8),DAG激酶(9),以及最近特征化MRCK(米肌强直性营养不良激酶-第页兴高采烈的C类dc42-绑定k个inase)家族(10). 这些C1中含有结构域的蛋白质,PKCs已经得到了最广泛的研究是重要的治疗靶点(11). 在药物中针对PKC的临床试验候选药物,如bryostatin 1和PEP005针对PKC的C1结构域,而不是其催化结构域现场。
经典的和新的PKCs在其N末端都含有调控蛋白结合DAG/佛波酯的区域串联C1结构域C1a和C1b(12). 多项研究试图定义这两个C1域在PKC中的各自作用监管,但问题仍不清楚。首字母在体外结合用传统PKC进行的测量表明,1摩尔的佛波酯每摩尔PKC的结合(13-15).另一方面,斯塔布斯等。,使用荧光佛波酯类似物,报道PKCα与每个PKC的两个配体结合(16). 此外,站点定向完整PKCα的C1a和C1b结构域的突变表明C1a和C1b结构域在对佛波醇12-肉豆蔻酸13-醋酸盐(PMA)和(-)辛基内酰胺V的反应(17). 同样,删除研究表明,PKCγ结合PDBu的C1a和C1b结构域同样具有高效力(三,18). 使用功能分析PKCα在酵母中表达,Shieh等。(19)删除单个C1并报告C1a和C1b在PMA刺激,任一缺失都会导致反应的效力,而对美泽兰来说,反应主要取决于C1a结构域,如果只存在C1b结构域,则响应要弱得多。使用孤立的C1域,Irie等。(20)建议C1aPKCα的结构域,但不包括PKCβ或PKCγ结合的结构域[三H] PDBu优先;不同的配体显示出普遍的模式相似,但选择性不同。使用合成的牛顿研究小组报道称,二聚双佛波醇(21)尽管两个C1PKCβII的结构域只针对潜在的膜相互作用生理背景下的一个C1结构域结合配体。
对于新型PKC,已经对PKCδ进行了许多研究研究孪生C1畴的等效性。的P11G点突变C1a结构域,它导致在孤立域(22),有对PKCδ的佛波酯依赖性易位几乎没有影响NIH3T3细胞,而C1b的相同突变导致佛波酯诱导易位的效力,表明佛波酯的主要作用佛波酯结合的C1b结构域(23). 的次要角色然而,C1a结构域被认为是因为C1a结构区也发生了突变因为C1b结构域导致了效力进一步7倍的转移。使用相同的C1a和C1b结构域的突变,Bögi等。(24)发现绑定PKCδC1a和C1b结构域的选择性似乎是配体依赖性。而PMA和吲哚生物碱吲哚内酰胺和辛基吲哚内酰胺选择性依赖于C1b结构域,选择性为美泽兰、12-脱氧山梨醇13-单酯前列腺素和12-脱氧山梨醇13-苯乙酸和大环内酯-苔藓抑制素1(24). 在在体外使用PKCδ,Cho群的孤立C1a和C1b结构域的研究(25)描述了两个C1结构域对DAG和佛波酯的亲和力相反;即这个C1a结构域与DAG的亲和力较高,C1b结构域的亲和力也较高对佛波酯的亲和力。通过以下方法未观察到选择性差异艾丽尔等。(20).
PKC已成为治疗癌症和其他情况,如糖尿病视网膜病变或黄斑变性(26-30).激酶抑制剂是一种很有前景的靶向PKC的方法恩扎他汀,一种对PKCβ具有中等选择性的抑制剂其他PKC亚型(但仍对其他一些非PKC激酶具有活性)是目前正在进行多项临床试验。药物的替代策略发展的目标是PKC的C1调控域。强有力的证据这种方法的原理是由多种天然产物提供的,例如bryostatin 1和PEP005也在临床试验中并且其指向C1结构域。这方面的潜在优势方法是同源靶点数量较少,<30个DAG-敏感C1与超过500个激酶相比的结构域,以及脂质环境的多样性提供了特异性,形成了配体与C1结构域结合的半衰期。因为不同的PKC亚型可能诱导拮抗活性,抑制一种亚型可能是功能等同于激活拮抗亚型(31).
和苯内酰胺一起(20,32),DAG内酯有为操纵配体提供了一个强大的合成平台:C1结构域相互作用(31). 对于例如,DAG内酯衍生物130C037在PKCα和PKCδ的重组C1a和C1b结构域以及RasGRP相对于PKCα的高度选择性(33). 同样,我们已经展示了改性的DAG内酯(二氧戊环酮)可以提供额外的点与PKCδC1b结构域结合的配体接触(34). 此类研究提供可以操纵配体-C1结构域相互作用以产生新的识别模式。可以通过以下方式获得进一步的选择性二价化合物,利用C1a和C1b域(35).更好地理解PKC中两个C1结构域的差异作用监管对于合理开发此类化合物至关重要。在这个通过分子操纵完整的C1a或C1b结构域进行研究PKCδ,我们发现C1a和C1b结构域在PKCδ调节。C1b结构域主要用于配体结合和用于整个PKCδ分子的膜移位。的C1a域完整的PKCδ在配体结合中仅起次要作用,但稳定质膜上用于下游信号传导的PKCδ分子。在此外,我们还证明了PKCδ的单个C1结构域的影响并不严重依赖于他们在监管领域的地位。
材料和方法
材料—[20-三H] 12,13-二丁酸佛波酯([三H] PDBu,17 Ci/mmol)购自PerkinElmer Life Sciences(马萨诸塞州波士顿)。PDBu和PMA来自LC实验室(马萨诸塞州沃本)。大脑磷脂酰丝氨酸(PS)和卵磷脂酰胆碱(PC)购自Avanti Polar Lipid(Alabaster,AL)。谷胱甘肽纯化试剂S公司-转移酶(GST)融合蛋白来自Pierce生物技术公司(伊利诺伊州罗克福德)。细胞培养基和试剂从Invitrogen获得。LB肉汤和细菌培养剂来自KD Medical,Inc.(马里兰州哥伦比亚)。DNA引物来自因维特罗根。小鼠抗GFP单克隆抗体购自Roche应用科学(印第安纳波利斯)。小鼠单克隆抗β-肌动蛋白抗体和抗鼠IgG购自Bio-Rad(加州大力士)。
野生型PKC的分子工程δ-GFP至生成不同的C1突变体-PKCδ的小鼠cDNA为如别处所述插入pEGFP-N1矢量(36). 三组突变在PKCδ-GFP质粒DNA上进行,以生成不同的C1突变体,如图所示图。1简而言之,GeneTailor™定点突变系统(Invitrogen)用于将限制性位点插入PKCδ-GFP模板符合制造商的协议。δC1a和δC1b片段来自野生型pDNA使用Platinum®通过PCR检测PKCδ-GFP(wt/PKCδ/GFP)Pfx公司DNA聚合酶(Invitrogen)。单个δC1a(C1a/PKCδ-GFP)和δC1b(C1b/PKCδ-GFP)突变体,双δC1a(C1a-C1a/PKCδ-GFP)和δC1b(C1b-C1b/PKCδ-GFP)突变体,以及位置转换的δC1a(C1a-2/PKCδ-GFP)和δC1b(C1b-1/PKCδ-GFP)突变体如补充材料。
图1。
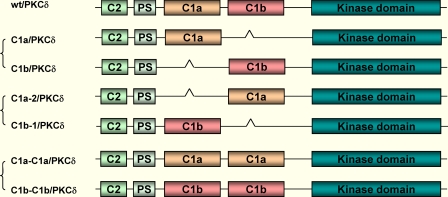
完整PKC C1突变体的结构δ. 野生型PKCδ在C端与GFP融合。单个δC1a或δC1b包含突变体(删除其中一个C1结构域)位置开关C1突变体(交换δC1a的位置和δC1b结构域),以及双δC1a或δC1b突变体(用一个C1结构域替换另一个)按所述制备在“材料和方法”下
荧光蛋白标记物的表达和成像PKC公司δ在活细胞中-LNCaP细胞(取自ATCC,Manassas,VA)在37°C下在含有10%的RPMI 1640培养基中培养胎牛血清、青霉素(50单位/ml)和链霉素(0.05 mg/ml)5%一氧化碳2增湿的大气。GFP融合蛋白的质粒使用Lipofectamine 2000(Invitrogen)转染细胞根据制造商的说明。的表达式转染24h后检测荧光蛋白。共焦荧光使用蔡司MRC1024共焦扫描头(蔡司,纽约州桑伍德)安装在尼康显微镜上,带有60×平面复消色差透镜如前所述(33).
细胞组分的制备及膜的Western Blot分析换位-CHO-K1细胞(取自弗吉尼亚州马纳萨斯州ATCC)接种在60-mm组织培养皿上并转染PKCδ-GFP或使用Lipofectamine 2000的所示C1域突变体。转染的转染后24小时用指定的药物处理细胞。之后药物处理,细胞单层用培养基清洗一次然后用冰镇的Dulbecco磷酸盐缓冲盐水清洗一次(KD Medical,Inc.,哥伦比亚,马里兰州)。用400μl20米米Tris-HCl(pH 7.4)加蛋白酶抑制剂混合物(Sigma)。然后在Eppendorf管中用一个每次脉冲6s。将200μl超声混合物转移至贝克曼超离心管,以200000×克在贝克曼超速离心机中分离细胞溶质和细胞膜分数如前所述(33). 膜移位PKCδ-GFP和C1突变体对配体处理的反应是由Western blotting测定细胞溶质分数与配体浓度的关系为量化。
免疫沉淀法纯化蛋白质-CHO-K1细胞接种在60mm的组织培养皿上。PKCδ-GFP质粒DNA并且使用Lipofectamine将C1突变体转染到CHO-K1细胞中2000(Invitrogen),根据制造商的说明。24小时后转染后,用冰片清洗细胞单层两次无钙磷酸盐缓冲盐水2+/镁2+和用400μl裂解缓冲液(1%Triton X-100 in)培养磷酸盐缓冲盐水和蛋白酶抑制剂)在冰上放置20分钟收集细胞并在4°C下以6000 rpm离心15 min。这个上清液通过添加小鼠进行免疫沉淀单克隆抗GFP抗体(Roche Applied Science)并在4°C,轻轻旋转3小时。30μl蛋白质A/G-琼脂糖然后将珠子(加州圣克鲁斯圣克鲁斯生物技术公司)添加到混合物,并在4°C下持续培养过夜。珠子当时是用1ml裂解缓冲液洗涤两次,用1ml 20ml洗涤一次米Tris-HCl(pH 7.5)加0.03%Triton X-100。最后,用珠子装订蛋白质悬浮在30μl的20m溶液中米三氯化氢+0.03%以下激酶的Triton X-100和[三H] PDBu绑定分析。
体外激酶分析-纯化蛋白的激酶活性PKCδ-GFP和C1突变体使用Pep-Tag®分析蛋白激酶C非放射性检测试剂盒(普罗米加,威斯康星州麦迪逊)根据制造商的说明。简单地说,上述5μl将含有PKCδ-GFP蛋白或C1突变体的珠子与不同浓度的PDBu(或PKC抑制剂GF109203X,EMD化学品(纽约州吉布斯敦)、特定肽底物和PKC反应缓冲液,包括PS:PC脂质混合物(200μg/ml磷脂酰丝氨酸和800μg/ml磷脂酰胆碱)30分钟摄氏度。将管子置于95°C加热条件下,停止反应封闭10分钟,然后在0.8%琼脂糖凝胶上在100℃下分离样品V持续7分钟。磷酸化肽向阳极(+)迁移,而非磷酸化肽向阴极(-)迁移。强度使用ImageJ软件(National美国卫生研究院(NIH)图片)。
[三H] PDBu结合分析-Scatchard分析在本研究中进行,以确定离解常数(K(K)d日值)用于[三H] PDBu绑定到纯化的野生型PKCδ-GFP蛋白和C1突变体其他地方(22). 竞争的如其他地方所述进行结合分析(37)以确定K(K)我某些弱亲和力突变体的值[三H] PDBu和测量甲泽兰和的结合亲和力1,2-二辛基甘油。
蛋白激酶C下调的蛋白质印迹分析δ由PMA提供在NIH3T3细胞中-NIH3T3细胞(取自弗吉尼亚州马纳萨斯州ATCC)在6孔组织培养皿中播种(Costar®、Corning Inc.、NewYork,NY)在Dulbecco改良的Eagle培养基(ATCC,Manassas,VA),含10%胎牛血清、青霉素(50单位/ml),以及5%CO中的链霉素(0.05 mg/ml)2大气。质粒DNA将野生型PKCδ-GFP和C1突变体转染到含脂蛋白2000的细胞。PKCδ-GFP和C1突变体水平通过Western blotting检测PMA处理的NIH3T3细胞。简单地说,24转染后h,用PMA处理细胞(0,10,100,1000,3000和10000 n米)在37°C下持续24小时。治疗后,细胞在RIPA缓冲液(50 m米特里斯(pH值7.5),150米米氯化钠、1%Triton X-100、0.1%十二烷基硫酸钠、10 mg/ml脱氧胆酸(钠盐)加蛋白酶抑制剂在冰上30分钟然后离心。收集上清液并进行在4-20%聚丙烯酰胺凝胶(Invitrogen)上进行SDS-PAGE,然后转移至聚偏二氟乙烯膜(Millipore,Billerica,MA)。老鼠单克隆抗GFP抗体(Roche Applied Science)和抗鼠IgG分别为用于检测GFP标记的PKCδ蛋白。膜也是小鼠抗β-肌动蛋白单克隆抗体作为印迹载体控件。ECL(Amersham)最终观察到免疫染色生物科学)。使用ImageJ扫描胶片并进行量化软件(NIH Image)。
结果
分离的C1a和C1b结构域的酚酯结合PKCδ
PKCδ的孪生C1结构域(即C1a和C1b)具有44%的序列同源性(图2A类).它们在PKCδ的调节区被一个22的连接子分隔开氨基酸。两个C1结构域包含相同的基本结合残基,形成有利的配体插入三级结构,这意味着它们可以平等地结合配体。然而,一个复杂的问题是C1域的功能可能会受到它们所处的环境的影响居住在PKC。这两个因素的相对重要性,C1域结构和上下文仍不清楚。此外,努力定义C1a和C1b结构域在配体识别和功能中的各自作用在完整的PKC内部产生了相互矛盾的答案,反映出PKC监管的复杂性(21,23,25). 这些问题非常重要与针对C1域的治疗药物设计策略的相关性(38).
图2。
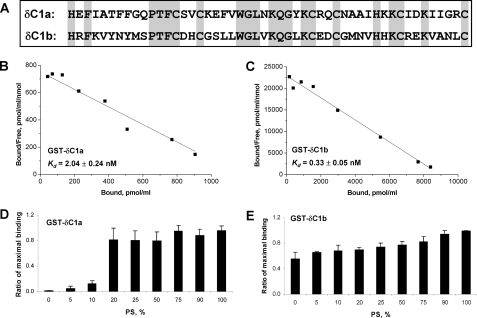
PKC C1a和C1b结构域的分离鉴定δ在体外。A类,比较δC1a和δC1b域。B类和C类、Scatchard图第页,共页[三H] PDBu与纯化和分离的δC1a和δC1b域。GST标记了δC1a和δC1b将结构域与浓度不断增加的[三H] PDBu公司在18°C下,在100μg/ml PS存在下保持10分钟使用聚乙二醇沉淀分析进行测量,如下所述“材料和方法。”点数代表平均值±一次代表性测定中三次测定的S.E实验。另外两个实验给出了类似的结果。这个K(K)d日表示为平均值±S.E(n个= 3独立实验)。D类和E类,的依赖项[三H] PDBu与GST标记的δC1a和δC1b结合分析中磷脂组成的结构域。在这些分析中,而不是分析中的100μg/ml PSB类和C类,的该分析含有100μg/ml总磷脂,包括以下混合物PS:PC,其中指示了PS:PC混合物中PS的重量%的值。的特定绑定[三H] PDBu(2-5个米)到C1不同PS:PC比例(0,5,10,20,25,50,75、90和100%的PS),并绘制为每个分析中的最大结合。这个酒吧在每个图中表示三个独立实验的平均值。误差线,±S。E。
为了进一步阐明这些问题,我们首先比较了效力两个C1结构域作为佛波酯结合的分离片段在里面体外单个C1结构域,从全长PKCδ中提取通过PCR,标记GST并从BL-21(DE3)中纯化大肠杆菌属大肠杆菌如其他地方所述(33). Scatchard分析用于测量结合亲和力(K(K)d日值),共个单个δC1a和δC1b域[三H] PDBu.公司。在含有100μg/ml磷脂酰丝氨酸、,这是我们通常的化验条件。δC1a和δC1b均显示出高绑定亲缘关系[三H] PDBu,由δC1a结合领域(K(K)d日=2.04±0.24牛顿米)谦逊地比δC1b域弱(6倍)(K(K)d日=0.33±0.05牛顿米) (图2,B类和C类).
与结合亲和力的有限差异相比PS要求的显著差异[三H] PDBu绑定PKCδ的C1a和C1b结构域。对于这些实验[三H] PDBu公司在固定浓度(100μg/ml)的总磷脂,具有可变百分比的PS和脂质的剩余部分as PC。结合是相对于分析中的最大结合来表达的。作为如所示图2D类,的δC1a结构域对PS有显著依赖性,最大值<20%当PS的总磷脂百分比低于时结合20%,并且当PS不存在并且所有磷脂都是PC时几乎没有任何结合。相反,δC1b结构域对PS的依赖性小得多[三H] PDBu结合,在缺乏PS,其中所有磷脂都是PC(图。2E类). PS优先定位于质膜。δC1a和δC1b对PS用于[三H] PDBu绑定意味着这两个C1域可能有利于配体相互作用的不同亚细胞位置。
PKCδC1a和C1b结构域的移位福尔波酯
接下来,我们比较了PKCδ的单个C1结构域在完整的细胞。将隔离的δC1a和δC1b结构域与GFP。GFP标记的结构体随后在LNCaP细胞中表达佛波酯PMA的细胞内分布和反应共焦显微镜观察(图。三). 表达了两个结构的类似级别在相同条件下进行可视化。两个C1结构域显示不含佛波酯时细胞内分布不同。δC1a主要定位于细胞核(图3A类),而δC1b主要分布在胞浆中,一些与内膜(图。三B类). 两个C1结构域均快速出现(3分钟内)1μ米PMA,但随着时间的推移,易位模式的稳定性表现出了戏剧性的变化差异。δC1a结构域向质膜的移位是应用PMA后持续(图3A类),而δC1b结构域仅短暂停留在质膜上(图3B类). 作为PMA在细胞内平衡后,δC1b结构域迅速从细胞内质膜呈斑块状分布。因为细胞持续灌入含有PMA的介质、击穿或PMA随时间的损失不能解释瞬态质膜易位。同样,在这段时间内,佛波酯不会导致GFP结构的分解。GFP本身对PMA没有反应。
图3。
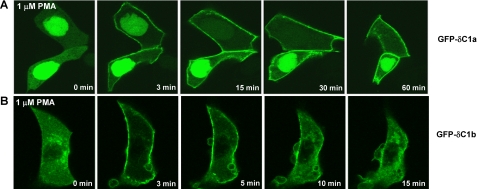
GFP标记的C1a的移位(A类)和C1b(B类)PKC域δ作为对活LNCaP细胞中PMA的反应时间的函数。表达GFP标记的C1a或C1b结构域的细胞用1μ米项目管理局。荧光的重新分布用蔡司MRC1024共焦显微镜监测蛋白质的功能添加PMA后的时间。每隔15或30秒拍摄一次图像面板代表三个独立的典型示例实验。
易位的动态差异可能由两个C1结构域对PS配体相互作用的不同依赖性,如如上文所述在体外结合分析(图2,D类和E类). 添加后,佛波酯不会立即在细胞内平衡。相反,佛波酯首先分为质膜只随后分布到内部膜,因为它继续平衡(39). 因为稳健δC1a与佛波酯的结合需要PS的存在在PMA存在的情况下,δC1a将移向质膜。之后然而,PMA已经进一步平衡,因此PMA现在既存在于在质膜和内膜中,δC1a不会由于内膜缺乏,因此对内膜进行重新平衡足够的PS。因为δC1b的结合对PS的依赖性小得多,δC1b将移动以反映细胞膜中的PMA。
C1a和C1b结构域的微分作用的表征在完整PKCδ中
分离C1片段的结果表明,C1a和PKCδ的C1b结构域对佛波酯具有较强的结合亲和力。在以下实验中,我们有兴趣探究完整PKCδ中的两个C1结构域。
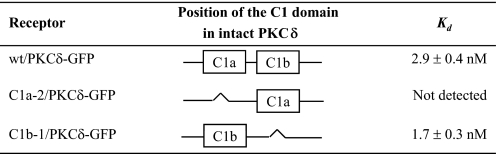
完整PKCδ,单个C1域的删除指示δC1b域具有更高的效力苯酚酯体外结合δC1a公司域-我们首先使用PKCδ-GFP质粒DNA作为模板删除δC1a或δC1b域以研究缺失对配体结合和细胞膜移位的影响PKCδ-GFP(图1). 这个野生型PKCδ-GFP蛋白和单个δC1a和δC1b突变体(指定为C1a/PKCδ-GFP和C1b/PKCδ-GFP以表示保留的C1结构域)在CHO-K1细胞中表达,并用用抗GFP单克隆抗体进行免疫沉淀,如下所述“材料和方法”[三H] 然后测定纯化蛋白的PDBu。单身δC1a和δC1b突变体在结合效力[三H] PDBu公司(表1). 当δC1b该结构域被删除,保留δC1a结构域,PKCδ显示结合力显著降低(45倍)[三H] PDBu公司(K(K)d日=132±13牛顿米),与野生型(K(K)d日=2.9±0.4牛顿米). 在相反,删除δC1a结构域不会影响全部;单个δC1b突变体显示K(K)d日值(2.8±0.5牛顿米)与野生型非常相似。这个结果与隔离δC1a和δC1b的情况大不相同结构域,其对[三H] PDBu公司(图2,B类和C类).
表1。
的比较在体外结合亲和力(K(K)值)野生型PKCδ-GFP和单个C1a和C1b突变体不同配体
GFP标记的构建物在CHO-K1细胞中表达。蛋白质是然后用抗GFP抗体免疫沉淀制备。Scatchard试验(用于K(K)d日值)和竞争性结合分析(用于K(K)我值),以测量结合效力这些蛋白质的配体。数值表示为平均值±S.E。用于三个独立的实验。
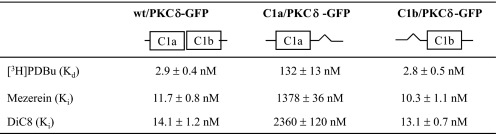
确定结果可能反映以下行为的程度我们检测了特定的配体,测量了单个配体的结合亲和力其他两种配体的δC1a和δC1b突变体,mezerein和DiC8。它之前有报道称,美泽兰在诱导PKCδ易位的PKCδC1a和C1b结构域在NIH 3T3细胞中(24).同样,据报道,DAG可能优先选择C1a结构域,与佛波酯对C1b结构域的偏好形成对比(16,40). 与此相反据报道,我们的C1结构域缺失突变体显示出与δC1a和δC1b的mezerein或DAG衍生物DiC8域和PDBu一样。mezerein和DiC8的结合力都弱得多单个δC1a突变体的亲和力(在K(K)我分别为值)与野生型相比,而对于单个δC1b,它们的结合效力保持不变突变体(表1).
PKC公司δ响应时未能转移到膜上当δC1b域已删除-进一步研究C1a和C1b结构域在配体反应性中的作用完整的PKCδ,我们评估体内单体易位PKCδ-GFP的δC1a和δC1b缺失突变体。质粒编码PKCδ-GFP的C1a或C1b结构域缺失突变体在LNCaP细胞中表达。突变体在细胞内的重新分布然后用共焦显微镜监测对PMA的反应。所有细胞均为在类似条件下观察,对单个细胞进行检查显示出相似水平的构建体表达。总体水平瞬时转染中构建物的表达没有差异>20%(参见补充图S4)。C1域删除显示标记为PMA对PKCδ膜转运的差异效应。在缺少δC1a结构域,δC1b/PKCδ-GFP(只有δC1b结构域)首先转移到质膜,然后转移到核膜对1μ的响应米项目管理局(图4B类). 然而,它的质膜移位是非常短暂的,不像野生型PKCδ-GFP(图。4A类). 它在质膜上停留不超过10min,然后移回电池内部。这些易位动力学与孤立δC1b结构域的动力学非常相似(图3B类). 上另一方面,与孤立的C1a结构域相比,在缺乏δC1b结构域单个δC1a突变体(C1a/PKCδ-GFP)没有显示PMA浓度达到10的任何膜移位μ米(图。4C类). 此外,这种缺乏体内回应是不依赖配体。易位对C1b结构域的类似依赖性DiC8和mezerein处理的细胞均可见(补充图S1和S2)。
图4。
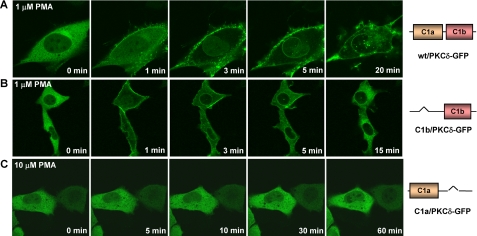
野生型PKC的易位δ-GFP和单个C1活性LNCaP细胞中对PMA反应的结构域突变体。表达用PMA处理GFP标记的野生型或突变型PKCδ。这个用蔡司MRC1024监测荧光蛋白的重新分布共焦显微镜作为添加PMA后时间的函数。图像每30秒就被抓获一次。A类,野生型时间序列图像添加1μ后的PKCδ-GFP米项目管理局。B类,时间PKCδ-GFP的单个δC1b突变体的序列图像(带有δC1a域删除)添加1μ米项目管理局。C类,单个δC1a突变体的时间序列图像添加10后的PKCδ-GFP(δC1b结构域缺失)μ米项目管理局。每个面板表示来自三个独立的实验。
C1b结构域在配体识别中的主要作用用C1a和C1b缺失突变体证明是完全一致的我们早期发表的实验(34)其中我们引入了PKCδC1a或C1b结构域的第27位突变,改变了Gln变成Glu。尽管这种突变几乎没有(C1a)或(C1b)变化在C1结构域的整体结构中,对于孤立的C1a结构域导致佛波酯或DAG结合亲和力显著丧失。对于隔离的PKCδ的C1b结构域,该突变差异性地导致二氧杂蒽酮(一种具有额外结合力的DAG衍生物)的亲和力与Gln侧链酰胺的相互作用),但对佛波酯或DAG结合。C1a结构域的突变对易位,除此之外,有一个适度的转变,支持转移到内膜。相反,C1b结构域突变二氧杂蒽酮引起的移位受阻,而保留了对佛波酯或DAG的反应,提供了强有力的控制PKC的整体功能没有被突变破坏。
C1a或C1b结构域在完整PKC中的位置δ做不影响其对配体的结合活性-删除其中一个C1结构域当然可能导致完整蛋白质的构象变化,这可能有助于对剩余配体的不同反应δC1a或δC1b结构域,因为其在完整结构中的位置发生了改变蛋白质。我们之前的突变研究,如上所述,提供了一种方法来解决这个问题。
为了探索C1结构域的位置和功能之间的关系,我们研究C1结构域的位置对配体结合的影响PKC。为此,我们切换了δC1a或PKCδ-GFP单C1结构域缺失突变体中的δC1b结构域(C1a/PKCδ-GFP或C1b/PKCδ/GFP)至δC1b或δC1a域分别为(图1和表2)然后测量它们的结合亲和力[三H] PDBu公司在体外如图所示在里面表2,更改定性位置对结合力影响不大。我们仍然检测到位置开关δC1b突变体具有很高的结合亲和力(C1b-1/PKCδ-GFP)(K(K)d日= 1.7 ± 0.3n个米),与野生型相比(K(K)d日=2.9±0.4牛顿米). 然而,我们无法检测到位置开关δC1a的任何结合活性在检测的配体范围内发生突变。
表2。
体外位置开关C1突变体的结合亲和力PKC的δ-GFP用于[三H] PDBu与野生型
值表示为三个独立值的平均值±S.E实验。
这个体内易位分析证实δC1结构域对佛波醇的敏感性没有重大影响酯类。所有细胞在类似条件下均可见对表现出类似结构表达水平的细胞进行检测。这个位置切换的单个C1突变体在瞬时转染差异不超过15%(重复的平均值实验,数据未显示)。就像之前的单个δC1b突变(C1b/PKCδ-GFP),位置开关单个δC1b突变体(C1b-1/PKCδ-GFP)仍表现为快速但短暂的质膜1μ的易位反应米项目管理局(图5A类),而位置开关的单个δC1a突变体(C1a-2/PKCδ-GFP)仍然存在即使PMA浓度增加到10,也没有任何反应μ米为了进一步探讨这一点,我们还生成了一个C1a/C1b位置反转全长PKCδ突变体(C1b-C1a/PKCδ-GFP)看看C1结构域的位置对其功能的影响。我们发现在完整的PKCδ中反转C1a和C1b的位置分子对佛波酯PKCδ的效力没有任何影响二者都在体外和体内(未显示数据)。总之,我们的结果表明,在任何位置,δC1a域单独PKCδ不能被佛波酯转运到膜我们的条件。当δC1b单独处于任一位置时,PKCδ-GFP可以被转运但在质膜上不稳定。
图5。
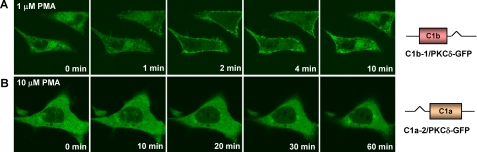
PKC的位置开关C1突变体的易位δ对活LNCaP细胞中的PMA作出反应。表达用PMA处理PKCδ-GFP的位置开关C1突变体。这个用蔡司MRC1024监测荧光蛋白的重新分布共焦显微镜作为添加PMA后时间的函数。图像每30秒捕获一次。A类,位置切换的时间序列图像单个δC1b突变体(δC1b位于正常位置δC1a)添加1μ米项目管理局。B类,时间位置切换的单个δC1a突变体的序列图像(具有δC1aδC1b的正常位置)μ米项目管理局。每个面板表示来自三个独立的实验。野生型PKCδ-GFP的对照(未显示数据)与图4A类。
PKC对苯酚酯敏感性的影响δ-GFP时间两个C1域都具有δC1a或δC1b类结构-除了产生C1-缺失突变体以研究C1a和C1b结构域在PKCδ激活中的作用,我们还生成双C1a(C1a-C1a/PKCδ-GFP)和双C1b(C1b-C1b/PKCδ-GFP)突变体(图。1)研究替换C1结构域之一的效果另一个关于完整PKCδ蛋白对配体的敏感性互动。我们做到了在体外像往常一样与这些药物进行结合分析双δC1a或δC1b突变体以比较它们的结合亲和力具有相应的单个δC1a和δC1b突变体。结果结果很有趣。当δC1a域被替换时对于δC1b结构域(产生双δC1b区域)完整PKCδ-GFP的亲和力保持不变(K(K)d日=2.3±0.3牛顿米)作为野生型(K(K)d日=2.9±0.4牛顿米)和单个δC1b突变体(K(K)d日= 2.8 ± 0.5n个米) (表3).有趣的是,尽管双δC1a突变体(δC1b被δC1a取代)到PDBu仍然保持较低(K(K)d日=11.5±1.1牛顿米)比野生型PKCδ(K(K)d日= 2.9 ± 0.4n个米),其效力明显(10倍)高于单个δC1a突变体(K(K)d日= 131.8 ± 12.8n个米) (表3)或第个,共个位于C1b结构域通常占据位置的单个C1a突变体(表2). 这增加了双δC1a突变体的敏感性也显示在在里面活泼地易位分析(图。6). 在类似条件下观察所有细胞,并且对表现出类似结构水平的单个细胞进行检测表达。The levels of overall expression of the constructs in the瞬时转染差异不超过1.8倍(平均2倍实验,数据未显示)。而单个δC1a突变体没有显示对10μ米项目管理局(图6A类),双人δC1a突变体表现出对10微米米尽管膜的重新分布有点缓慢且范围缩小(图。6B类). 该结果证实了δC1a结构域完整的PKCδ确实维持了配体结合的结构,并且可能在某些情况下有助于配体结合。
表3。
比较在体外单个C1a的结合亲和力和PKC的双C1a(或C1b)突变体δ-GFP用于[三H] PDBu公司
值表示为三个独立值的平均值±S.E实验。
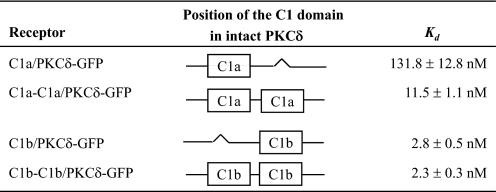
图6。
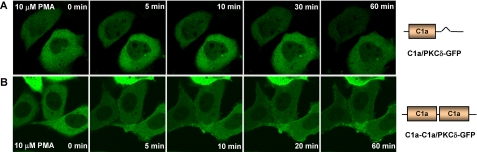
单链C1a突变体和双链C1a突变的易位PKC公司δ-活LNCaP细胞中GFP转PMA。表达单个δC1a(只有一个δC1a域)和双δC1a用PMA处理PKCδ-GFP(含两个δC1a结构域)突变体。用蔡司MRC监测荧光蛋白的重新分布添加PMA后,1024共焦显微镜随时间变化。每30秒拍摄一次图像。A类,单个的时间序列图像添加10μ米项目管理局。B类,添加10后双δC1a突变体的时间序列图像μ米项目管理局。每个面板代表的典型示例三个独立的实验。
虽然双C1b突变体表现出类似的结合能力佛波酯在体外与野生型和单个C1b突变体,显示出几个差异体内首先双C1b突变体在无PMA的LNCaP细胞(参见补充图S3)。其次双C1b突变体表现出持续的质膜移位反应至1μ米LNCaP细胞中的PMA(补充图S3),而单个C1b突变体是暂时的。
的角色δC1a和δ中的C1b域调节PKC的下调δ由PMA提供-两者都有的分析在体外绑定和用于体内易位证明δC1b结构域是完整PKCδ中的配体识别。我们想确定这是否就下游功能后果而言,情况仍然如此。长期(24h) PMA的治疗(41). 我们因此研究了δC1a和δC1b结构域在PMA诱导PKCδ-GFP下调。野生型PKCδ-GFP单个和双C1突变体在NIH3T3细胞中表达24小时。然后用不同浓度的PMA处理细胞24小时用抗GFP蛋白对总细胞裂解物进行Western blot分析抗体。代表性结果如所示图7A类。谱带强度的量化如图所示图7B类如预期,PMA后野生型PKCδ-GFP呈剂量依赖性下调治疗。单δC1a(C1a/PKCδ-GFP)和双δC1a(C1a-C1a/PKCδ-GFP)突变体在蛋白质水平上没有任何变化经PMA治疗后。这与我们之前的结果一致,没有δC1b结构域,单个δC1a和双δC1aPKCδ-GFP突变体对PMA不敏感或在灵敏度。与PMA的强大敏感性相一致双δC1b(C1b-C1b/PKCδ-GFP)突变体也表现出明显的剂量依赖性下调对PMA的反应。这些结果再次表明δC1b在驱动配体相互作用和PKCδ活化。单个δC1b突变体(C1b/PKCδ-GFP)对佛波酯没有表现出下调反应。相反,它在PMA浓度为1μ米和3μ米我们检查了单个δC1b的表达水平在没有PMA的情况下转染24和48小时后突变,发现该突变体的表达非常不稳定。与野生型相比和单个δC1a突变体,单个δC1b突变体的水平为转染48小时后明显减少(补充图S4)。我们也有检查双δC1b突变体的表达。其表达水平在此期间非常稳定(未显示数据)。这似乎是合理的PKCδ的单个C1b突变体的表达明显增强因此表示在佛波酯存在下的稳定性。
图7。
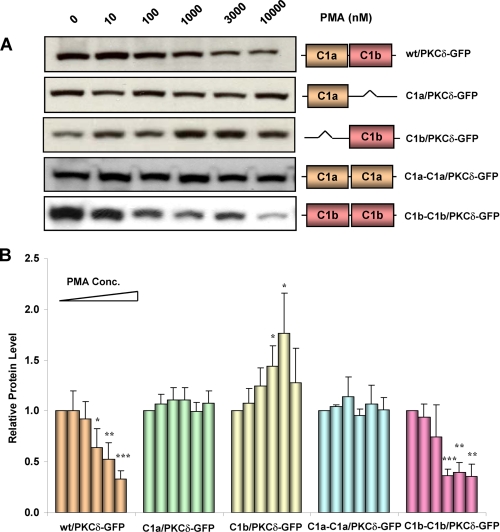
野生型下调的Western blot分析PKC公司δ-GFP及其单、双C1a和C1b突变体NIH3T3细胞对PMA的反应。野生型过度表达细胞PKCδ-GFP,以及单个和双C1突变体与增加浓度(0 n米,10牛顿米, 100n个米, 1 μ米, 3 μ米、和10μ米)然后收集细胞,用RIPA缓冲液,并进行Western印迹。对于每个构造,相等在对照组和处理过的样品中加入一定量的蛋白质。A类,结果来自一个单一的、有代表性的实验。每个实验进行了四到五次,结果相似。B类,作为PMA功能的蛋白质水平变化的量化浓度。对胶片进行扫描,相对带强度为使用ImageJ确定。未处理条带的强度为标准化为1。每个柱在中图表代表四到五次实验的平均值。统计显著性(与每组中对照样本的值)t吨测试:*,第页< 0.05;**,第页< 0.01; 和***,第页< 0.001.
单、双C1a或C1b突变体的体外激酶检测PKC公司δ-GFP公司-我们表演了在体外激酶用C1突变体进行分析,以进一步研究PKCδ激活中的C1a和C1b结构域。单个和双C1a或C1bPKCδ-GFP突变体在CHO-K1细胞中表达和纯化抗GFP抗体免疫沉淀。然后纯化的蛋白质评估在体外PDBu诱导的激酶活性使用Promega试剂盒(PepTag®非放射性检测蛋白激酶C)。结果总结如下图8; 基础活性野生型PKCδ-GFP标准化为1.0。的激酶活性PDBu对野生型PKCδ-GFP的刺激作用剂量依赖性方式。单个δC1a(C1a/PKCδ-GFP)(图8A类)也不是双δC1a(C1a-C1a/PKCδ-GFP)(图8B类)突变体对PDBu的反应显示出增强的激酶活性。缺乏回应进一步证实了在没有δC1b结构域的情况下,δC1a结构域单独不能识别配体并调节PKCδ酶。
图8。
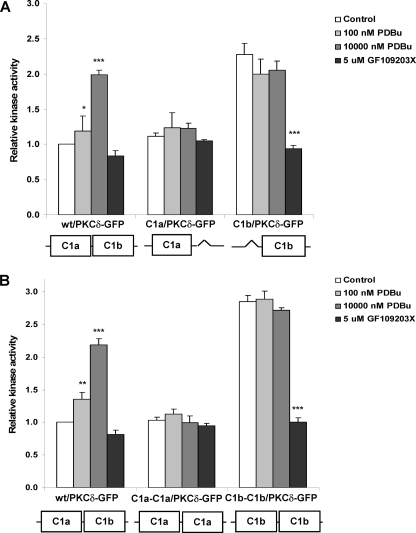
体外野生型PKC激酶测定δ-GFP公司还有单打和双打δC1a和δC1b突变体。野生型PKCδ-GFP和C1突变体在CHO-K1细胞中表达,用于24小时,表达水平相似。使用用抗GFP抗体免疫沉淀,然后等量在30°C下与不同浓度的PDBu孵育30分钟。这个在体外用Promega蛋白激酶测定激酶活性C符合制造商协议的检测试剂盒。激酶活性如中所示图表相对于基础活性进行计算野生型PKCδ-GFP。A类,比较在里面体外野生型PKCδ-GFP和单个PKCδ-GFP的激酶活性δC1a和δC1b突变体与PDBu浓度和PKC抑制剂GF109203X对小鼠脑组织基底激酶活性的影响构造。B类,比较在体外激酶活性野生型PKCδ-GFP和双δC1a和δC1b突变体作为PDBu浓度和PKC抑制剂作用的函数GF109203X对构建物的基础激酶活性的影响。这个酒吧在里面每个图表表示三个独立变量的平均值实验。误差线,±S。E.统计意义(与每组对照样本的值进行比较)t吨测试:*,第页< 0.05;**,第页< 0.01; 和***,第页< 0.001.
与易位结果相反,PDBu没有进一步刺激单个δC1b(C1b/PKCδ-GFP)的激酶活性(图8A类)或双δC1b(C1b-C1b/PKCδ-GFP)(图8B类)突变体。相反,在缺乏PDBu的情况下,单个δC1b的基础活性并且双δC1b突变体已经达到了与受激wt/PKCδ。我们证实了高基础激酶活性PKC抑制剂GF109203X可显著降低PKC图8),表明这高基础激酶活性不是来自其他污染激酶用PKC进行铜净化。这些结果表明,如果没有δC1a域,PKCδ起作用在体外好像是佛波酯刺激。
讨论
以PKC的C1域为目标是一种互补且有前途的策略在众多丝氨酸/苏氨酸中获得PKC同工酶的选择性具有同源催化位点的激酶(31). 使用DAG类似物Marquez小组已经开发出具有结合亲和力的化合物接近佛波酯(31)并且具有选择性C1域之间(33). 这个当前的研究试图探索理性设计背后的两个相关问题C1靶向配体。首先,C1a和配体反应中PKCδ的C1b结构域?两个C1域的存在性通过二价体为PKC活性的复杂调节提供了机会配体(35). 然而,尽管其中一些配体的性能比对于单体,这与合作结合的预期不一致到两个C1域(21,35). 第二,什么是C1结构域对配体响应的相对贡献每se与C1结构域在PKCδ内的位置?Irie和同事们建议,合成的C1结构域可以代表一种强健的选择性配体发现平台(20). 另一方面,我们例如,已经表明,130C037对C1a的相对选择性PKCα和PKCδ的C1b结构域在其完整PKC的选择性(33).
在本研究中,通过分子工程对完整的C1结构域进行研究PKCδ,我们检测了孪晶δC1a和δC1b的作用配体结合、膜移位和酶激活中的结构域PKCδ。PKCδ的C1a和C1b结构域的序列为44%相同,以及锌结合残基和配体必需的残基结合几乎是完全保守的(图2A类). 这个表明这两个C1结构域的主干结构非常类似的,以及我们之前的静态结构的计算机建模δC1a和δC1b在这两个领域(34). 这个δC1a的结合裂宽了~1º,δC1a结构域不像δC1b结构域那样深入膜(34). 我们的结果来自本研究中孤立的δC1a和δC1b结构域与这些计算机建模结果一致。作为孤立的碎片δC1a和δC1b结构域对[三H] PDBu绑定在体外(图2,B类和C类)和PMA诱导的质膜移位在里面活泼地(图3). 然而,我们也发现于在体外结合分析δC1a域到[三H] PDBu完全依赖于PS,而在缺乏PS的情况下,δC1b结构域的结合基本上保持不变(图2,D类和E类). 这可以解释我们的体内结果,其中GFP-δC1b仅通过项目管理局。类似PMA的嗜脂性佛波酯首先在血浆中积聚膜,然后重新平衡到内膜中(39). 因为δC1b在没有PS、GFP-δC1b的情况下,结构域可以与佛波酯结合预计将反映PMA随时间和当PMA从血浆中平衡到内膜时薄膜。另一方面,因为δC1a与佛波醇的结合酯类依赖于PS(主要位于质膜上),因此,GFP-δC1a仅限于PS所在的质膜以及PMA(图。三A类).
相同的解释适用于删除的δC1a的行为PKCδ-GFP突变体(产生单个δC1b突变体)。类似在完整的PKCδ中,当δC1a结构域缺失,该响应不依赖于特异配体。PKCδ的单个C1b突变体表现出类似的模式佛波酯PMA的瞬时易位反应(图4B类),发送给DAG类似物DiC8(图S1)和瑞香烷类似物甲泽兰(图S2)。这个表明δC1a结构域的重要功能之一可能是稳定质膜上的酶,使其能够接触血浆膜定位底物。
在我们的研究中,δC1b结构域的缺失(产生单个δC1a突变体)导致PKCδ-GFP与配体(表1,图4、和补充图S1和S2),与δC1b域播放一致结合配体和驱动细胞膜移位的主要作用整个PKCδ分子。这一结果与其他发现一致。对于例如,通过将第11位的脯氨酸残基突变为甘氨酸(P11G)在完整PKCδ的C1a或C1b结构域中,Szallasi等。发现δC1b结构域在易位中起主要作用PKCδ对PMA的响应(23). 对此的确认结论是以二氧杂蒽酮为配体得出的(34). 对于此类DAG衍生物,PKCδ中的Q185E突变导致二氧杂蒽酮的C1b结构域,而非佛波酯或DAG;类似地,PKCδ的Q185E突变体的易位在对二氧杂蒽酮,但不适用于佛波酯或DAG。中相应的突变相反,PKCδ的C1a结构域未能阻断对任何配体的反应。特殊强度二恶英酮-Q185E体系对PMA和DAG的结合活性为保留,以积极控制二氧杂蒽酮的结合损失C1b结构域的功能并没有发生更大的全球性变化失去响应。
使用双功能(二聚体)佛波醇衍生物和PKCδC1a或C1b结构域中脯氨酸11的突变,牛顿组也有证明δC1b结构域在配体中起主要作用绑定(21). 然而,与δC1a或δC1b结构域的相同突变,Bogiet(等)阿尔。说明C1a和C1b域选择性为配体依赖性,至少对于易位(24). 鉴于PMA和吲哚生物碱吲哚内酰胺和辛基内酰胺选择性依赖于在C1b结构域中,未观察到甲泽兰的选择性12-脱氧山梨醇13-单酯前列腺素或12-脱脱氧山梨醇13-苯乙酸,或大环内酯bryostatin 1(24). 斯塔赫林等。也表明PKCδ的C1a和C1b结构域具有相反的功能DAG与佛波酯的亲和力;即δC1a结构域是DAG优先选择性和δC1b结构域显示优先性佛波酯(25). 一个导致不同情况下不同反应模式的因素可能是支持配体相互作用的脂质环境的差异以及其他共同激活剂的可变贡献。因此相同配体的相对选择性与苔藓抑制素1,对于相同的PKC亚型,PKCα与PKCδ,显示小鼠角质形成细胞的显著差异(42)和小鼠3T3细胞(43). 无论如何,我们没有观察δC1b结构域选择性对配体的依赖性本研究采用删除一个或其他C1结构域的方法。这可能意味着未链接的C1b域仍在为膜相互作用。
应该注意的是,除配体结合外的其他功能建议用于C1a域。例如,最近的研究使用了PKCβ揭示PKCβ的C1a结构域可能与新的E3相互作用连接酶RINCK(环-与对照inase),促进PKC的下调(44). 同样,PKCδ为受酪氨酸磷酸化的调节,以及这些调节之一酪氨酸残基位于187位,位于C1a结构域(45).
单个δC1a和δC1b突变体的不同敏感性配体结合的(C1a/PKCδ-GFP和C1b/PKCδ-GFP)可能无法可以用蛋白质因缺失而发生的结构变化来解释,因为双δC1a和δC1b突变体(C1a-C1a/PKCδ-GFP和C1b-C1b/PKCδ-GFP)反映了它们的行为(表3). 此外佛波酯的特性和灵敏度再次保持不变无论单个C1结构域是否位于其正常位置或交换到其他C1域的位置(表2和图5). 我们的结论是主要是C1结构域本身,而不是PKCδ中的特定位置决定其活性的因素。
C1b结构域在PKCδ调节中的主要作用也是对于下调和激酶活性的功能性测量明显。在没有δC1b结构域的情况下,无论是一个还是两个C1aPKCδ中存在结构域,酶不能PMA下调体内(图7)或通过PDBu激活在体外(图8). 这个单个δC1b突变体(C1b/PKCδ-GFP)在下调分析是唯一的。蛋白质并没有被下调单个δC1b突变体的水平随着PMA剂量的增加而增加(图7). 这可能反映了在缺乏PMA的情况下,这种结构的表达水平较低,可能反映出它已经处于一种构造中,因此容易受到降解(补充图S4)。它强调了PKC异构体的处理,反映配体对PKC的两种作用异形体本身及其与细胞内其他元素的相互作用(46). 我们的激酶分析证明纯化的单个和双δC1b突变体(C1b/PKCδ-GFP和C1b-C1b/PKC-δ-GPP)显著升高基础激酶活性在体外(图8). 这表明δC1a结构域有助于维持PKC处于非活性状态,可能是通过稳定催化位点中的假底物区域。在在任何情况下,我们都没有观察到诸如ERK磷酸化或凋亡等影响响应单个δC1b或双δC1b的过度表达细胞中的突变体。
本研究观察到双δC1a突变体(C1a-C1a/PKCδ-GFP)表现出改善(尽管仍然非常微弱)佛波酯与单个δC1a突变体的敏感性比较(C1a/PKCδ-GFP)(表3和图6). 这表明δC1a结构域维持了配体结合的结构完整的PKCδ。然而,由于δC1a与配体的相互作用要求质膜上存在PS(图2D类),它必须首先被带到质膜上进行高亲和力的相互作用。δC1b可能在使C1a结构域更接近血浆方面发挥作用膜和配体。然后,与C1a域的交互可能加强PKCδ分子在等离子体中的定位薄膜。
使用二价配体利用C1之间差异的方法包含域的目标将寻求利用两个C1域的存在在经典和新型PKC和PKD中,与单个C1域相比在其他目标中,以及在经典PKC、新型PKC和PKD。方法的先决条件,当然,这两个C1结构域都可以识别配体。我们在这里的证据,这表明PKCδ的C1a结构域是活跃的,尽管较少potent为二价配体方法提供了支持。此外表明孪生C1结构域的相对作用可能不同于不同的PKC亚型,虽然超出了本文的范围,暗示了进一步的选择性机会。
*这项工作全部或部分得到了美国国立卫生研究院院内研究程序,NCI癌症中心研究。这篇文章的出版费用是支付部分页面费用。因此,本条必须特此标记“广告“根据18《美国法典》第1734节仅用于说明这一事实。
脚注
2使用的缩写是:PKC,蛋白激酶C;PDBu,佛波12,13-二丁酸盐;PMA,佛波醇12-肉豆蔻酸酯13-醋酸盐;二C8,1,2-二辛醇甘油;绿色荧光蛋白;CHO,中国仓鼠卵巢;PS、,磷脂酰丝氨酸;磷脂酰胆碱;GST、谷胱甘肽S公司-转移酶;二酰甘油;E3,泛素蛋白异肽连接酶。
工具书类
-
1Nishizuka,Y.(1992)《科学》258607-614[内政部] [公共医学] [谷歌学者]
-
2Ron,D.和Kazanietz,M.G.(1999)FASEBJ.十三1658-1676[公共医学] [谷歌学者]
-
三。Ono,Y.、Fujii,T.、Igarashi,K.、Kuno,T.、Tanaka,C.、Kikkawa,U.和Nishizuka,Y.(1989)Proc。国家。阿卡德。科学。美国。南澳大利亚864868-4871[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
4Zhang,G.、Kazanietz,M.G.、Blumberg,P.M.和Hurley,J.H。(1995)细胞81 917-924[内政部] [公共医学] [谷歌学者]
-
5Rozengurt,E.、Sinnett-Smith,J.和Zugaza,J。(1997)《生物化学》。社会事务处理。25 565-571[内政部] [公共医学] [谷歌学者]
-
6Ahmed,S.、Lee,J.、Kozma,R.、Best,A.、Monfries,C.和Lim,L。(1993)《生物学杂志》。化学。26810709-10712[公共医学] [谷歌学者]
-
7Brose,N.、Rosenmund,C.和Rettig,J.(2000)货币。操作。神经生物学。10 303-311[内政部] [公共医学] [谷歌学者]
-
8Ebinu,J.O.、Bottorff,D.A.、Chan,E.Y.、Stang,S.L.、Dunn,R。J.和Stone,J.C.(1998)《科学》2801082-1086[内政部] [公共医学] [谷歌学者]
-
9Topham,M.K.(2006)《细胞杂志》。生物化学。97474-484[内政部] [公共医学] [谷歌学者]
-
10Choi,S.H.、Czifra,G.、Kedei,N.、Lewin,N.E.、Lazar,J.、Pu、,Y.、Marquez,V.E.和Blumberg,P.M.(2008)J。生物化学。28310543-10549[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
11Hofmann,J.(2004)货币。抗癌药物目标4125-146[内政部] [公共医学] [谷歌学者]
-
12Newton,A.C.(2001)《化学》。版次1012353-2364[内政部] [公共医学] [谷歌学者]
-
13Hannun,Y.A.和Bell,R.M.(1986)J。生物化学。2619341-9347[公共医学] [谷歌学者]
-
14Kikkawa,U.、Takai,Y.、Tanaka,Y.,Miyake,R.和Nishizuka,Y。(1983)《生物学杂志》。化学。25811442-11445[公共医学] [谷歌学者]
-
15Mosior,M.和Newton,A.C.(1996年)生物化学351612-1623[内政部] [公共医学] [谷歌学者]
-
16Slater,S.J.、Ho,C.、Kelly,M.B.、Larkin,J.D.、Taddeo,F.J.、。,Yeager,M.D.和Stubbs,C.D.(1996)《生物学杂志》。化学。2714627-4631[内政部] [公共医学] [谷歌学者]
-
17Bogi,K.、Lorenzo,P.S.、Acs,P.、Szallasi,Z.、Wagner,G.S.和Blumberg,P.M.(1999)FEBS Lett。456 27-30[内政部] [公共医学] [谷歌学者]
-
18Burns,D.J.和Bell,R.M.(1991)J。生物化学。26618330-18338[公共医学] [谷歌学者]
-
19Shieh,H.L.、Hansen,H.、Zhu,J.和Riedel,H。(1995)分子致癌物。12 166-176[内政部] [公共医学] [谷歌学者]
-
20Irie,K.、Nakagawa,Y.和Ohigashi,H.(2004)货币。药物设计。101371-1385[内政部] [公共医学] [谷歌学者]
-
21Giorgione,J.、Hysell,M.、Harvey,D.F.和Newton,A.C。(2003)生物化学4211194-11202年[内政部] [公共医学] [谷歌学者]
-
22Kazanietz,M.G.,Wang,S.,Milne,G.W.,Lewin,N.E.,Liu,H.L。,和Blumberg,P.M.(1995)J.Biol。化学。27021852-21859[内政部] [公共医学] [谷歌学者]
-
23Szallasi,Z.、Bogi,K.、Gohari,S.、Biro,T.、Acs,P.和Blumberg,P.M.(1996)《生物学杂志》。化学。27118299-18301[内政部] [公共医学] [谷歌学者]
-
24Bogi,K.、Lorenzo,P.S.、Szallasi,Z.、Acs,P.、Wagner,G.S.和Blumberg,P.M.(1998)《癌症研究》。581423-1428[公共医学] [谷歌学者]
-
25Stahelin,R.V.,Digman,M.A.,Medkova,M.,Ananthanaarayanan,B。,Rafter,J.D.、Melowic,H.R.和Cho,W.(2004)J。生物化学。27929501-29512[内政部] [公共医学] [谷歌学者]
-
26Teicher,B.A.(2006)临床。癌症决议125336-5345[内政部] [公共医学] [谷歌学者]
-
27Griner,E.M.和Kazanietz,M.G.(2007)Nat.Rev.癌症7281-294[内政部] [公共医学] [谷歌学者]
-
28Martiny-Baron,G.和Fabbro,D.(2007年)药理学。第55号决议477-486[内政部] [公共医学] [谷歌学者]
-
29Fields,A.P.和Gustafson,W.C.(2003)方法分子生物学。233519-537[内政部] [公共医学] [谷歌学者]
-
30Anderson,P.W.、McGill,J.B.和Tuttle,K.R。(2007)货币。操作。Nephrol公司。高血压。16 397-402[内政部] [公共医学] [谷歌学者]
-
31Marquez,V.E.和Blumberg,P.M.(2003)Acc.Chem.化学。第36号决议434-443[内政部] [公共医学] [谷歌学者]
-
32马赫,U.R.、列文,N.E.、布隆伯格,P.M.和科齐科夫斯基,A.P。(2006)化学。医药化学。1 307-314[内政部] [公共医学] [谷歌学者]
-
33Pu,Y.,Perry,N.A.,Yang,D.,Lewin,N.E.,Kedei,N.,Braun,D。C.、Choi,S.H.、Blumberg,P.M.、Garfield,S.H、Stone,J.C.、Duan,D.和Marquez,V.E.(2005)生物学杂志。化学。28027329-27338[内政部] [公共医学] [谷歌学者]
-
34Choi,Y.、Pu,Y..、Peach,M.L.、Kang,J.H.、Lewin,N.E.、Sigano、,D.M.、Garfield,S.H.、Blumberg,P.M.和Marquez,V.E。(2007)《医学化学杂志》。503465-3481[内政部] [公共医学] [谷歌学者]
-
35Nowak,I.(2007)货币。医学专题。化学。7355-362[内政部] [公共医学] [谷歌学者]
-
36Wang,Q.J.,Bhattacharyya,D.,Garfield,S.,Nacro,K.,Marquez,V.E.和Blumberg,P.M.(1999)《生物学杂志》。化学。27437233-37239[内政部] [公共医学] [谷歌学者]
-
37Wang,Q.J.,Fang,T.W.,Nacro,K.,Marquez,V.E.,Wang,S.和Blumberg,P.M.(2001)《生物学杂志》。化学。27619580-19587[内政部] [公共医学] [谷歌学者]
-
38Blumberg,P.M.,Kedei,N.,Lewin,N.E.,Czifra,G.,Pu,Y.,Yang,D.和Marquez,V.E.(2008)货币。药物目标9641-652[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
39Braun,D.C.、Cao,Y.、Wang,S.、Garfield,S.和Hur,G.MBlumberg,P.M.(2005)《摩尔癌症治疗》。4 141-150[公共医学] [谷歌学者]
-
40Slater,S.J.、Kelly,M.B.、Taddeo,F.J.、Rubin,E.和Stubbs,C.D.(1994)《生物学杂志》。化学。26917160-17165[公共医学] [谷歌学者]
-
41Choi,S.H.、Hyman,T.和Blumberg,P.M.(2006)癌症研究667261-7269[内政部] [公共医学] [谷歌学者]
-
42Szallasi,Z.,Denning,M.F.,Smith,C.B.,Dlugosz,A.A.,Yuspa,S.H.、Pettit,G.R.和Blumberg,P.M.(1994)摩尔。药理学。46840-850[公共医学] [谷歌学者]
-
43Szallasi,Z.、Smith,C.B.、Pettit,G.R.和Blumberg,P.M。(1994)《生物学杂志》。化学。2692118-2124[公共医学] [谷歌学者]
-
44Chen,D.,Gould,C.,Garza,R.,Gao,T.,Hampton,R.Y.,和Newton,A.C.(2007)《生物学杂志》。化学。28233776-33787[内政部] [公共医学] [谷歌学者]
-
45Brodie,C.和Blumberg,P.M.(2003)细胞凋亡819-27[内政部] [公共医学] [谷歌学者]
-
46Gould,C.M.和Newton,A.C.(2008)货币。药物靶点9614-625[内政部] [PMC免费文章] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。




