摘要
线粒体遗传病可由线粒体DNA(mtDNA)缺失、点突变或缺失形式的缺陷引起,最终导致氧化磷酸化的丧失。这些突变可能是自发的、母体遗传的,也可能是维持线粒体DNA的基因中遗传性核缺陷的结果。这篇综述的重点是我们目前对核基因突变的认识,这些核基因突变会导致线粒体DNA改变,并导致线粒体耗竭综合征(MDS)、进行性外眼肌麻痹(PEO)、共济失调神经病变或线粒体神经胃肠道脑肌病(MNGIE)。迄今为止,所有这些致病核基因都属于两类之一:其产物直接作用于线粒体DNA复制叉的基因,例如POLG、POLG2、和TWINKLE公司或其产物为线粒体提供DNA复制所需的脱氧核苷酸三磷酸池的基因,如TK2、DGUOK、TP、SUCLA2、ANT1可能还有新发现的MPV17型。
关键词:DNA聚合酶γ、核苷酸池、线粒体DNA缺失综合征、进行性外眼肌麻痹、共济失调神经病变
简介
线粒体疾病可由线粒体DNA(mtDNA)或编码线粒体功能蛋白质的核基因的遗传缺陷引起(1). 线粒体基因组包含37个基因,所有这些基因都直接或间接参与ATP的产生。其中13个基因编码参与电子传递的蛋白质亚基,以进行氧化磷酸化。其余24个基因编码线粒体蛋白质合成所需的转移RNA(22个基因)和核糖体RNA(2个基因)。线粒体DNA拷贝数高;一个单元格包含1000到10000个副本。MtDNA通过DNA聚合酶γ(polγ)的协同作用复制,其辅助亚基p55(由POLG2型)以及复制因子,如线粒体单链DNA结合蛋白和Twinkle解旋酶。Polγ是哺乳动物线粒体中唯一已知的DNA聚合酶,因此承担着DNA复制和DNA修复功能的负担(2). 自1999年以来,已有近十几个与线粒体耗竭综合征(MDS)和相关疾病相关的基因被发现(表1). MDS不仅包括常见的疾病,如进行性外眼肌麻痹(PEO)和共济失调,还包括一些非常罕见的三羧酸(TCA)循环异常。中的突变POLG、POLG2、TWINKLE、和ANT1型与PEO相关,编码线粒体核苷酸代谢相关酶的几个核基因突变可导致线粒体DNA点突变、缺失或缺失,从而导致线粒体综合征。线粒体严重依赖线粒体转运蛋白或补救途径酶来提供线粒体DNA复制所需的脱氧核苷酸三磷酸(dNTPs)。
表1。
| 基因 |
无序一
|
染色体座 |
功能 |
| POLG公司 |
PEO/阿尔卑斯山/共济失调 |
25年第15季度 |
线粒体DNA聚合酶 |
|
| POLG2型 |
PEO公司 |
23–24年第17季度 |
polγ辅助亚基 |
|
| 双胞胎(PEO1) |
PEO/共济失调 |
24年第10季度 |
线粒体DNA解旋酶 |
|
| ANT1型 |
PEO公司 |
34至35年第4季度 |
腺嘌呤核苷酸转运体 |
|
| TP(ECGF1) |
MNGIE公司 |
22季度13.32 |
胸苷回收 |
|
| DGUOK公司 |
MtDNA缺失 |
第2页,共13页 |
脱氧鸟苷激酶 |
|
| TK2型 |
MtDNA缺失 |
22年第16季度 |
线粒体胸苷激酶 |
|
|
脱氧核糖核酸b条
|
MCPHA公司 |
17季度25.3 |
脱氧核苷酸载体(焦磷酸硫胺转运) |
|
| MPV17型 |
线粒体DNA缺失 |
第21页至第23页 |
线粒体内膜蛋白 |
|
| SUCLA2公司 |
线粒体DNA缺失 |
13季度12.2–13季度13.3 |
琥珀酸-CoA连接酶 |
|
| RRM2B型 |
线粒体DNA缺失 |
8季度23.1 |
核糖核苷酸还原酶 |
DNA聚合酶γPOLG公司基因
由基因突变引起的疾病POLG公司,编码polγ催化亚单位的基因具有高度异质性(2–5). 中的突变POLG公司与多种疾病相关,如PEO、帕金森综合征、更年期提前、阿尔卑斯综合征、线粒体神经胃肠道脑肌病(MNGIE)、感觉共济失调性神经病、构音障碍和眼病(SANDO)(2,4–6). 在POLG公司(图1) (6)(另请参见人类聚合酶γ突变数据库,http://dir-apps.niehs.nih.gov/polg).
图1。
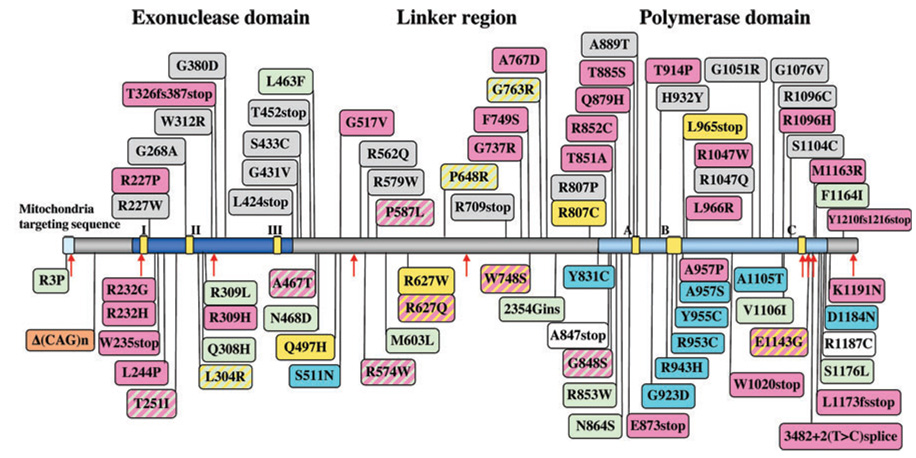
人类polγ蛋白示意图,显示致病氨基酸替换和多态性的位置。该蛋白在前三分之一处排列有核酸外切酶结构域,其次是长连接子区域,聚合酶结构域位于C末端第三。疾病替代由方框表示:浅蓝色方框,常染色体显性PEO;浅绿色框,常染色体隐性突变;灰色方框,零星PEO;粉红盒子,阿尔卑斯综合征;黄盒,共济失调神经病变样综合征;橙色盒子,男性不育。带条纹的盒子代表在多种疾病中发现的疾病替代。红色箭头表示非同义多态性氨基酸的变化。
POLG公司和进行性外眼肌麻痹
中的突变POLG公司于2001年首次被确定为PEO基因座(7). PEO是一种线粒体疾病,与线粒体DNA缺失和/或线粒体DNA突变和缺失积累有关(7–9). PEO的特点是发病较晚(18至40岁),双侧上睑下垂,眼球外肌进行性弱化,导致上睑下坠和眼睑麻痹。近端肌肉无力、消瘦以及运动不耐受也与PEO有关。PEO患者的骨骼肌呼吸链酶活性降低,并在病理上显示出粗糙的红色纤维。首次在可遗传常染色体显性PEO(adPEO)的意大利家系中发现肌肉活检分离出的线粒体DNA的多重大规模缺失(8).
迄今为止,除了一个例外,所有常染色体显性遗传POLG公司导致PEO的突变位于polγ的聚合酶域。其中两个替代物R943H和Y955C改变了与传入的dNTP直接相互作用的侧链(10). 这些酶保留<1%的野生型聚合酶活性,并显示加工能力严重下降。极低的催化活性和由此导致的DNA合成停滞是R943H和Y955C杂合子严重临床表现的两个最可能的原因(10). 在没有核外裂解校对的情况下,Y955C替代也会使核苷酸错误插入率增加10–100倍(11). 在为评估酵母中同源突变而开发的酵母模型中混合蛋白1基因,我们发现Y757C突变体(人类中的Y955C)表现出线粒体DNA异常,表明氧化损伤和非常高的微小频率(12). 在一项相关研究中,Baruffini等人表明,这种高微小频率可以通过抗氧化剂治疗或上调核糖核苷酸还原酶来挽救(13). 在Y955C转基因小鼠模型中POLG公司针对心脏,小鼠出现心肌病,线粒体DNA丢失,线粒体DNA中8-oxo-dG水平升高(14). 总之,这些表型表明,携带Y955C突变的患者可能具有较高的氧化损伤,并可能受益于抗氧化治疗。
帕金森综合征和adPEO在两名帕金森病患者中同时发生POLG公司(15). 帕金森综合征在最初的PEO症状出现几年后出现。具有PEO的女性POLG公司突变可能会导致更年期提前,并伴有高促性腺激素和低雌激素浓度,表明卵巢早衰(15,16).
大多数POLG公司突变与常染色体隐性PEO(arPEO)相关,PEO患者通常是具有两个不同突变PEO等位基因的复合杂合子。例如,在反式与其他人POLG公司PEO、共济失调神经病变和阿尔卑斯综合征的错义突变(17,18). 在两个家系中发现A467T突变为纯合子突变,与中年严重共济失调相关(19). 生化分析表明,A467T突变体polγ仅具有约4%的野生型聚合酶活性,对核酸外切酶的影响不大(20). 此外,A467T polγ蛋白不能与其辅助亚单位p55相互作用,这通常是高度加工DNA合成所必需的(20). 然而,A467T是一种常见的突变,存在于0.6%的比利时人口中(17).
POLG公司和阿尔卑斯综合征
阿尔卑斯综合征是一种罕见但严重的常染色体隐性MDS疾病,影响幼儿。在生命的最初几年里,患者表现出渐进性痉挛性四联症,渐进性大脑退化导致精神恶化和癫痫发作,皮层失明,耳聋,肝衰竭,最终死亡。Naviaux等人报道了一名阿尔卑斯病患者,其电子传递链功能降低、二羧酸尿、暴发性肝衰竭、顽固性癫痫和乳酸酸中毒导致42个月时死亡(21). 骨骼肌活检显示mtDNA含量降低至野生型水平的30%,但未检测到polγ活性(21). 的排序POLG公司这些家系中的基因显示在POLG公司将Glu873(GAG)转化为一个终止密码子,并在连接子区域的外切酶域之后进行杂合A467T替换(18). 具有E873终止突变的波尔γ信使核糖核酸通过无义介导的衰变从信使核糖核酸库中去除,导致POLG公司只含有A467T突变(22). 迄今为止,报告的Alpers关联数量POLG公司突变从46个不同的先证者上升到35个以上(23–25). 在所有情况下POLG公司阿尔卑斯病患者的突变是隐性的,许多相同的突变也可能导致arPEO。A467T突变通常被发现为arPEO中的复合突变,是最常见的Alpers突变,被发现为纯合子突变或杂合子突变与其他突变结合。
POLG公司和共济失调神经病变
中的突变POLG公司也可导致共济失调神经病变综合征,发病年龄在青少年早期至30岁晚期。这种共济失调,也称为线粒体相关共济失调综合征(MIRAS)(26)、脊髓-小脑共济失调-癫痫综合征(SCAE)或SANDO(19),由常染色体隐性突变引起POLG公司在受影响的个体中产生多个mtDNA缺失。症状包括周围神经病变、构音障碍、轻度认知障碍、不自主运动、精神症状、肌阵挛和癫痫发作。A467T突变纯合子的共济失调患者在青少年早期至晚期出现症状(19,27). SANDO患者也被发现有复合杂合突变,其中一个是A467T突变POLG公司其他的等位基因和R3P、L304R或R627W(17). 一名共济失调-肌病综合征患者显示其中一名患者存在A467T突变POLG公司其他基因中的等位基因和R627Q和Q1236H突变POLG公司等位基因(28). 其他共济失调患者被发现是杂合子,其中一个等位基因A467T突变,W748S在顺式其他等位基因E1143G突变。E1143G突变最初在4%的普通人群中被鉴定为单核苷酸多态性(SNP)(6). 然而,这种突变与其他基因突变的累积报道POLG公司线粒体疾病的突变表明E1143G可能会加剧疾病进程(5,29). W748S突变与E1143G结合被发现是共济失调的常见原因(26,27). 芬兰人群的单倍型分析显示W748S突变的携带者频率为1:125,该等位基因在古代欧洲人群中具有共同的遗传起源(26).我们发现W748S突变单独导致聚合酶的催化活性低和严重的DNA结合缺陷(30). E1143G替代部分地挽救了W748S突变的有害影响,并似乎调节了疾病突变的影响POLG公司。
POLG公司和男性不育
人类POLG公司该基因包含一个10单位的CAG三核苷酸束,编码成熟蛋白N末端附近的聚谷氨酰胺延伸(31). 虽然CAG重复序列的缺失对组织培养细胞的线粒体功能没有明显影响(32)一些研究表明,CAG重复序列的改变与精子质量的下降有关,并导致欧洲人口中5%–10%的男性不育病例(33,34). 与这些研究相反,两项独立研究未能证实多态性CAG重复序列与POLG公司和男性不育(35,36).
POLG2型,POLγ的附属子单位
最近,编码副亚单位的基因发生了一次突变,POLG2型,在一名adPEO患者中报告(37). 这种突变导致G451E在不参与p55二聚化的环区发生取代。p55的重组G451E突变体的特征表明,突变的辅助亚基在与polγ催化亚基结合方面存在缺陷,并且不能刺激过程性DNA合成。未能增强催化亚单位的处理能力将导致复合体在DNA复制过程中停滞,这与在PEO中检测到的mtDNA缺失的积累一致。
线粒体DNA螺旋酶TWINKLE公司或人1基因
线粒体DNA解旋酶由TWINKLE公司基因,也称为PEO1公司与噬菌体T7 gp4螺旋酶相关。该基因首次作为第10、C10或f2号染色体上的PEO位点被分离(38). 衍生的氨基酸序列与T7 gp4解旋酶的C末端具有显著的序列同源性,但缺少T7 gp 4中发现的关键的引物相关序列(38,39). 筛选TWINKLE公司adPEO患者中与多个线粒体DNA缺失相关的基因,在12个受影响的家族中鉴定出11种不同的编码区突变,与该疾病共同分离(38). 大多数突变位于C端解旋酶结构域和N端区域之间的连接子区域。该区域被认为与单体之间的亚单位相互作用有关,从而形成功能性六聚体。
中的突变TWINKLE公司主要与adPEO有关,但一份报告描述了隐性TWINKLE公司突变是SANDO的病因(40). 小鼠转基因模型过度表达了多种PEO突变TWINKLE公司概述了人类PEO的许多特征,包括多个线粒体DNA缺失、进行性呼吸功能障碍和细胞色素c氧化酶缺乏(41).
腺嘌呤核苷酸转位体,ANT1型
ANT1是发现的三种腺嘌呤核苷酸转运体蛋白之一,是线粒体内跨膜蛋白,是线粒体中最丰富的蛋白质。ANT1作为一种由30-kDa单体组成的同型二聚体发挥作用,在心脏、肾脏、肝脏和骨骼肌中高度表达。其主要功能是将ATP从线粒体基质中运输出来,以交换ADP。考科宁等人(42)在一个adPEO家族中使用位置克隆将adPEO的基因座缩小到4q,包括ANT1型以及其他64个基因。的序列分析ANT1型在5个散发性adPEO家族和1名患者中,发现了两个杂合错义突变ANT1型(43). 常染色体突变A114P和散发突变V289M均位于蛋白质结构的跨膜结构域内。酵母同源基因中的类似A114P突变,美国陆军指挥控制中心,导致呼吸系统缺陷(43). 杂合T293CANT1型在一个希腊adPEO家族中发现突变(44),一名意大利散发性PEO患者的杂合V289M突变(45)以及德国adPEO家系中的杂合A90D突变。在德国家系中,尽管微卫星标记显示该等位基因是显性的,并且是从母亲那里遗传来的,但该患者血液中没有携带突变,表明存在种系嵌合体(46).
酵母基因中其他几种等效突变的表达美国陆军指挥控制中心在中澳大利亚航空公司2缺陷单倍体酵母菌株在非发酵碳源上造成了明显的生长缺陷,并降低了细胞呼吸(47). 这个美国陆军指挥控制中心与野生型相比,致病性突变体在ADP与ATP转运方面表现出明显缺陷。
胸腺嘧啶磷酸化酶,ECGF1型
胸苷磷酸化酶(TP)是胸苷和磷酸转化为胸苷和脱氧核糖-1-磷酸所需的嘧啶补救途径的一部分(图2). 中的缺陷ECGF1型,编码TP的基因,导致血液中胸腺嘧啶和尿嘧啶的积累。由于线粒体严重依赖于生成线粒体内dNTP池的补救途径,线粒体吸收多余的胸苷,从而刺激线粒体中胸苷激酶2合成多余的脱氧胸苷三磷酸(dTTP)。由此产生的线粒体脱氧核苷酸库失衡会导致线粒体DNA耗竭、多重缺失和点突变。
图2。
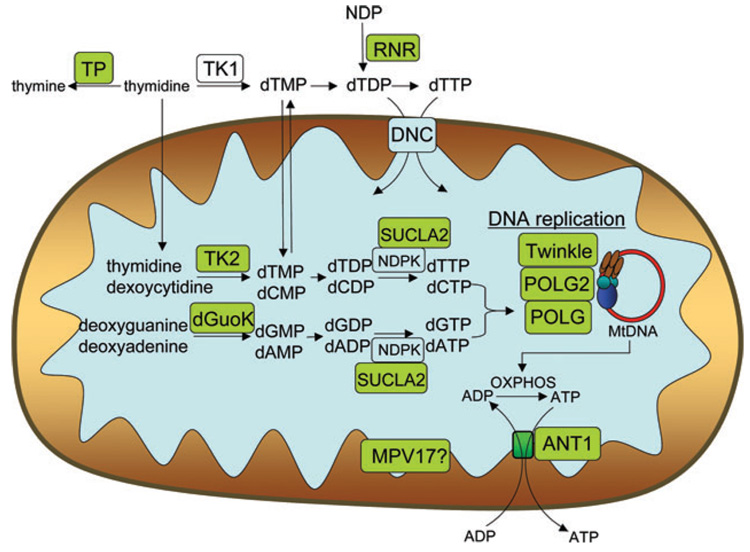
线粒体示意图,显示了当线粒体受到破坏时导致线粒体DNA突变或耗竭的酶途径。与MDS或mtDNA突变相关的基因产物位于绿色框中。
MNGIE是一种常染色体隐性遗传疾病,由ECGF1型(48,49). 到目前为止ECGF1型已知与MNGIE有关(49). 与PEO一样,该病与线粒体DNA的多重缺失和缺失有关(9). 发病年龄通常在20到50岁之间,典型的临床特征包括上睑下垂、PEO、胃肠动力障碍、恶病质、周围神经病、肌病和白质脑病(50,51). TP缺乏导致循环脱氧胸腺嘧啶浓度增加(52)和脱氧尿苷(53). 这些增加导致线粒体脱氧核糖核酸三磷酸池失衡,其作用可增加线粒体DNA突变(54).
在MNGIE患者组织中发现的80%以上的mtDNA突变是T到C的转变,之前有短时间的As(55). 这种特征性突变表明,更常见的错误插入T:dGMP(脱氧鸟苷单磷酸)导致了“next-nucleotide效应”(56)由于MNGIE细胞线粒体中TP缺乏导致dTTP浓度升高,这一现象迅速扩大(55). 此外,增加的胸腺嘧啶核苷衍生的dTMP(脱氧胸苷单磷酸)浓度升高可以抑制polγ的外切核酸酶活性(57). 在添加50µM胸腺嘧啶的培养基中生长的HeLa细胞显示出线粒体DNA缺失,dTTP和dGTP的线粒体池升高,这一结果重述了MNGIE中的许多遗传效应(54). 这些结果支持了一种突变机制,即dGTP和dATP之间的竞争与模板T相反(55).
一些令人兴奋的研究途径显示出治疗MNGIE的潜力。血液透析已被证明能暂时降低血液中的胸苷水平(58). 异基因干细胞移植在恢复TP活性和降低血浆胸苷水平方面取得了一些成功(59). 此外,反复输注血小板可以降低MNGIE患者血液中的胸苷水平(60).
线粒体胸腺嘧啶激酶,TK2型
在一项针对四名婴儿期致命性肌病和线粒体耗竭综合征的无关患者的研究中,Saada等人(61)发现线粒体呼吸链功能降低,肌肉中线粒体DNA数量减少。胸苷激酶2的序列分析(TK2型)该基因识别出两个纯合突变H90R或I181D中的任何一个。这些患者线粒体裂解物的活性分析显示TK2活性降低(61). 其他研究小组的DNA序列已经鉴定出总共14个错义突变,这些错义突变要么是隐性纯合子,要么是复合杂合子突变,以及来自12个肌肉无力和张力减退先证者的一个终止密码子(62–67). 线粒体dNTP池通过细胞溶质dNTP的主动转运或通过两种线粒体脱氧核苷激酶TK2和脱氧鸟苷激酶的作用而形成的补救途径产生(图2). 在非分裂细胞中,细胞溶质TK1和dNTP合成被下调,迫使线粒体dNTP池合成的负担转移到两个线粒体脱氧核苷激酶上。TK2型突变主要影响肌肉组织,对肝脏、大脑、心脏或皮肤没有影响。不同组织中TK2活性相对于线粒体DNA或细胞色素c氧化酶活性的定量有助于解释TK2缺乏的组织特异性(68).
脱氧鸟苷激酶,DGUOK公司
脱氧鸟苷激酶是另一种线粒体脱氧核苷激酶,将嘌呤核苷磷酸化为核苷酸单磷酸(图2). 在患有肝脑型MDS的三个近亲家系中进行纯合子作图,确定了染色体2p13上的一个区域,该区域包括脱氧鸟苷激酶基因,DGUOK公司(69). 该基因的序列分析确定了与疾病分离的核苷酸缺失(204delA)(69). 在21名MDS患者的筛查中,Salviati等人(70)发现三名患者(14%)患有DGUOK公司突变。这三名患者的表型差异很大,其中一名患者出现肝衰竭,但对肝移植反应良好(70). 为了确认DGUOK公司Wang等人表示,突变确实破坏了酶的功能(71)用这些取代物表征重组脱氧鸟苷激酶。他们发现R142K变异体对脱氧鸟苷(dG)的活性很低,而对脱氧腺苷(dA)没有活性。E227K蛋白对dG和dA具有正常的亲和力,但催化效率低,表明这种突变破坏了催化作用而不影响底物结合。C末端截断的变体是不活跃的。进一步分析发现,一名患有MDS、肌肉无力和严重线粒体肌病导致的运动不耐受的患者在DGUOK公司(65). 重组L250S-dGuoK蛋白的检测显示,与野生型酶相比,其活性小于1%,脱氧胞苷(dC)和脱氧胸腺嘧啶(dT)作为底物之间存在差异竞争(65). Freisinger等人(72)在六名患有婴儿肝性脑病和MDS的儿童中,还发现了五种新的突变和两种先前描述的突变。迄今为止,已有13种突变在DGUOK公司基因,大多数表现为来自14个先证者的纯合子突变(67,72).
脱氧核苷载体,SLC25A19型
2002年,Kelley等人(73)描述了宾夕法尼亚州兰开斯特县老阿米什族人的一种代谢紊乱,其特征是严重的先天性小头畸形、严重的2-酮戊二酸尿,通常在第一年内死亡。阿米什致命小头症(MCPHA)是一种常染色体隐性遗传疾病,在这一人群中,其发病率异常高,至少为1/500(73). 罗森博格(Rosenberg)等人通过对23个祖先相关家族进行系谱分析并结合全基因组扫描(74)定位了MCPHAt染色体17q25的基因。该区域包含线粒体脱氧核苷酸载体基因(脱氧核糖核酸或SLC25A19型)发现该基因含有一个错义突变,将Gly177变为Ala,但在252条控制染色体中未发现该基因。G177A突变体的功能分析挪威船级社证实突变蛋白的转运活性有缺陷(74).
A类挪威船级社到第12天,敲除小鼠导致100%的产前死亡,伴有神经管闭合缺陷和α-酮戊二酸升高(75). 然而,鼠标挪威船级社−/−细胞没有显示出mtDNA水平的任何降低。此外,线粒体核糖和脱氧核苷三磷酸水平正常,表明核苷酸转运可能不是DNC的主要作用。在酵母中发现一种具有类似氨基酸同源性的蛋白质,Tpc1p,可以将焦磷酸硫胺(ThPP)运输到线粒体中,以交换磷酸硫胺(76). 事实上,体外转运试验证实G177A挪威船级社没有导致核糖核苷酸或脱氧核苷酸进入线粒体的缺陷。此外,小鼠敲除的线粒体没有检测到ThPP,MCPHA细胞的线粒体ThPP水平降低。因此,ThPP水平的降低导致α-酮戊二酸二氢酶复合物无法正常发挥作用,从而导致这些患者体内α-酮戊二酸水平升高。
琥珀酰可可合成酶,SUCLA2公司
2005年报道了一个与线粒体DNA缺失相关的常染色体隐性脑肌病穆斯林小家系(77). 全基因组连锁图谱确定了13号染色体上的一个20-Mb区域,通过线粒体输入预测程序将该区域缩小为三个基因。其中之一的DNA测序SUCLA2公司基因第6外显子3′端出现43nt纯合缺失。这种突变导致受影响患者的mRNA第6外显子和第7外显子的部分缺失(77). 琥珀酸CoA合成酶是一种线粒体基质酶,在三羧酸(TCA)循环中催化琥珀酸-CoA和ADP可逆合成琥珀酸和ATP(或GTP)。SUCLA2公司编码琥珀酰辅酶A合成酶的β亚基。反向反应发生在克雷布斯循环中,而正向反应可能产生琥珀酰辅酶A,用于激活酮体和血红素合成。
根据这一初步发现,在法罗群岛人群中发现了12名常染色体隐性遗传线粒体脑肌病和甲基丙二酸升高患者(78). 甲基丙二酸升高是甲基丙二酸血症(MMA)的特征,这是一组异质性疾病,症状包括呕吐、脱水、嗜睡、癫痫发作、发育迟缓、进行性脑病。甲基丙二酸含量的增加会阻碍维生素B12转化为其两种代谢活性形式。积累的琥珀酰基CoA抑制甲基丙二酰CoA向琥珀酰基CoA的代谢,导致甲基丙二酰基Co A和MMA的积累。突变分析发现一种新的剪接位点突变SUCLA2公司导致外显子4的跳过。创始人效应导致法罗群岛人口的高发病率(1700人中有1人),携带者频率为1/33(78). 在一项相关研究中,意大利南部和法罗群岛14名患者的DNA测序证实SUCLA2公司所有患者的突变,并导致鉴定出三种新的突变(79). 法罗群岛人群中这些突变等位基因的频率为2%,对应于估计的1:2500纯合子频率(79).
柠檬酸循环的缺陷和甲基丙二酸的积累是如何导致线粒体DNA耗竭的尚不清楚。然而,免疫沉淀实验发现琥珀酰辅酶A合成酶与线粒体核苷酸二磷酸激酶复合物(80). Elpeleg等人(77)建议SUCLA2公司破坏与核苷酸二磷酸激酶的关联,导致核苷酸二磷酸酶在线粒体dNTP补救的最后一步中出现缺陷,从而导致dNTP减少和随后的mtDNA耗竭。在早期/新生儿发作脑肌病、肌张力障碍、耳聋和主要影响壳核和尾状核的类Leigh MRI异常患者中,应考虑琥珀酰CoA基因突变(79).
MPV17型
线粒体DNA不稳定性疾病列表中最新添加的基因位点之一是MPV17型基因(81). Calvo等人使用增强的计算算法来识别线粒体靶向基因产品(82)确定了1080个基因产物,其中包括368个先前预测不会定位于线粒体的基因产物。其中8个新基因,包括MPV17型,被确定为线粒体疾病的候选基因座(82). 在随附的一篇文章中,Spinazzolla等人(81)报告了几个患有MDS的家庭,其中患者在生命早期出现肝衰竭。全基因组连锁分析将该基因映射到与MPV17型DNA序列分析确定了Arg50的一个常见突变,在这些患者中突变为Gln或Trp。最初推测MPV17蛋白定位于过氧化物酶体,但人类和猴细胞的共焦免疫荧光显示MPV17与线粒体共定位(81). 一项平行研究在6名纳瓦霍神经性肝病患者中发现了相同的R50Q突变(83). 纳瓦霍神经肝病是美国西南部纳瓦霍发现的一种常染色体隐性多系统疾病。其特征是肝衰竭、严重感觉神经病变、角膜麻醉和瘢痕形成、脑白质脑病、发育不良和酸中毒(83以及其中的参考)。
这个MPV17型酵母中的同源物,SYM公司位于线粒体内膜。SYM1型缺失或突变SYM1型产生R51Q或R51W替代物(相当于人体中的R50Q或R50W),导致呼吸不足和线粒体DNA重排(81).MPV17型−/−小鼠出现年龄依赖性听力损失和肾小球硬化(84)同时肝脏、肌肉、大脑和肾脏中线粒体DNA减少,呼吸功能下降(81). 尽管MPV17的功能尚待阐明,但通过整合基因组分析发现该蛋白为鉴定MDS相关的其他基因铺平了道路。就像中的缺陷蔗糖2,中的缺陷MPV17型可能导致与核苷酸代谢蛋白的不稳定蛋白复合物,或膜相关蛋白-DNA核仁复合物的不稳定,其中mtDNA复制有望发生。
P53诱导核糖核苷酸还原酶,M2B马来西亚令吉
通过核糖核苷酸还原酶将核糖核苷二磷酸还原为脱氧核糖核糖核苷二磷酸,可以直接获得DNA复制所需的核苷酸前体。核糖核苷酸还原酶由两个亚基组成:一个大催化亚基R1和一个小R2亚基。细胞有两种形式的R2亚单位:一种是在S期最大表达的细胞周期调节形式,另一种是p53诱导形式,称为p53R2。p53R2形式是非增殖细胞中基本水平的DNA修复和mtDNA合成所必需的。最近,Bourdon等人(85)确定了RRM2B型编码p532B的基因是一个肌肉mtDNA严重缺失家族的全基因组连锁分析中的候选疾病基因。的序列分析RRM2B型在这个家族和其他三个受影响的家族中,发现了无义、错义和剪接位点突变以及在RRM2B型在控制染色体中未发现的基因。线粒体DNA严重缺失RRM2B型中断也在Rrm2b中得到证实−/−小鼠,证明该基因在线粒体DNA核苷酸代谢和线粒体疾病中的重要作用(85).
结论
线粒体DNA稳定性疾病可追溯到线粒体DNA复制的核心蛋白或与线粒体DNA复制所需的线粒体核苷酸前体供应有关的基因(图2). 中的突变POLG公司基因与几种线粒体疾病有关,包括致命的儿童疾病,如阿尔卑斯综合征、PEO、共济失调性神经病,也可能是男性导致失活和/或截短的蛋白质,这些蛋白质是POLG公司和TWINKLE公司PEO中常见的“显性负性”蛋白质会干扰其野生型对应物。许多参与核苷酸补救途径和核苷酸转运的基因与线粒体疾病有关,这一事实表明,不平衡的核苷酸库对线粒体DNA复制有害。小鼠模型、酵母遗传学和突变蛋白的体外生化分析对于理解这些基因中可遗传突变的体内后果具有重要价值。
总结要点
线粒体DNA复制疾病主要分为两类,直接影响DNA复制蛋白的疾病和影响线粒体核苷酸库的疾病。
这些疾病可影响多种器官,发病年龄不定。线粒体DNA复制疾病的具体表现通常由这些基因组内的突变类型决定。
许多症状通常与发病年龄和组织特异性有关。例如,肝病通常发生在生命早期,而骨骼肌肌病出现在生命后期。然而,这些患者似乎随时都会发生神经病变。
POLG公司在这些疾病中经常发生变异。
未来问题
更好地了解这些疾病的发病机制及其组织特异性。
需要开发合适的动物模型来模拟线粒体DNA复制疾病,以阐明组织表现和病理学。
开发可能减缓这些疾病进展的治疗方法。
致谢
作者感谢Rajendra Prasad、Jeffrey Stumpf和Rajesh Kasiviswanathan对本手稿的批评性阅读。这项审查得到了NIH国家环境健康科学研究所内部基金的支持。
- 线粒体DNA
线粒体DNA
- 波尔γ
DNA聚合酶γ,编码POLG公司基因
- MDS公司
线粒体DNA缺失综合征
- PEO公司
进行性外眼肌麻痹
- TWINKLE公司
线粒体解旋酶基因编码
- 数字NTP
脱氧核苷酸三磷酸
- MNGIE公司
线粒体神经胃肠脑肌病
- 桑多
感觉性共济失调性神经病、构音障碍和眼病
- 广告
常染色体显性
- 应收账
常染色体隐性遗传
- TP(转移定价)
胸苷磷酸化酶
脚注
披露声明作者不知道任何可能影响本次审查客观性的偏见。
引用的文献
-
1华莱士特区。人和鼠的线粒体疾病。科学。1999;283:1482–1488. doi:10.1126/science.283.5407.1482。[内政部][公共医学][谷歌学者]
-
2Graziewicz MA、Longley MJ、Copeland WC。线粒体DNA复制和修复中的DNA聚合酶γ。化学。2006年修订版;106:383–405. doi:10.1021/cr040463d。[内政部][公共医学][谷歌学者]
-
三。Tzoulis C、Engelsen BA、Telstad W等。A467T和W748S POLG突变引起的临床疾病谱:26例研究。大脑。2006;129:1685–1692年。doi:10.1093/brain/awl097。[内政部][公共医学][谷歌学者]
-
4DiMauro S,Davidzon G,Hirano M.多态性聚合酶。大脑。2006;129:1637–1639. doi:10.1093/brain/awl169。[内政部][公共医学][谷歌学者]
-
5Hudson G,Chinnery PF。线粒体DNA聚合酶γ与人类疾病。嗯,分子遗传学。2006;15(规范编号2):R244–R252。doi:10.1093/hmg/ddl233。[内政部][公共医学][谷歌学者]
-
6Longley MJ、Graziewicz MA、Bienstock RJ等。人类DNA聚合酶γ突变的后果。基因。2005;354:125–131. doi:10.1016/j.gene.2005.03.029。[内政部][公共医学][谷歌学者]
-
7Van Goethem G、Dermaut B、Lofgren A等。POLG突变与以mtDNA缺失为特征的进行性外眼肌麻痹相关。自然遗传学。2001;28:211–212. doi:10.1038/90034。第一个发现POLG公司是PEO的一个基因座,这导致了其他疾病的发现POLG公司突变。
-
8Zeviani M、Servedei S、Gellera C等。一种从D-loop区域开始线粒体DNA多重缺失的常染色体显性疾病。自然。1989年;339:309–311. doi:10.1038/339309a0。[内政部][公共医学][谷歌学者]
-
9Hirano M、Marti R、Ferreiro-Barros C等。基因组间通讯缺陷:导致线粒体DNA多重缺失和缺失的常染色体疾病。塞明。单元格。开发生物。2001;12:417–427. doi:10.1006/scdb.2001.0279。[内政部][公共医学][谷歌学者]
-
10Graziewicz MA、Longley MJ、Bienstock RJ等。常染色体显性遗传进行性外眼肌麻痹患者线粒体DNA聚合酶的结构-功能缺陷。自然结构。分子生物学。2004;11:770–776. doi:10.1038/nsmb805。[内政部][公共医学][谷歌学者]
-
11Ponamarev MV、Longley MJ、Nguyen D等。与进行性外眼肌麻痹相关的DNA聚合酶γ活性位点突变导致容易出错的DNA合成。生物学杂志。化学。2002;277:15225–15228. doi:10.1074/jbc。C200100200。[内政部][公共医学][谷歌学者]
-
12Stuart GR、Santos JH、Strand MK等。酿酒酵母线粒体DNA缺陷与DNA聚合酶γ突变与进行性眼外肌麻痹相关。嗯,分子遗传学。2006;15:363–374. doi:10.1093/hmg/ddi454。[内政部][公共医学][谷歌学者]
-
13Baruffini E、Lodi T、Dallabona C等。线粒体DNA聚合酶突变诱导的酿酒酵母表型的遗传和化学拯救,与人类进行性外眼肌麻痹相关。嗯,分子遗传学。2006;15:2846–2855. doi:10.1093/hmg/ddl219。[内政部][公共医学][谷歌学者]
-
14Lewis W、Day BJ、Kohler JJ等。MtDNA缺失、氧化应激、心肌病和转基因心脏靶向人类突变聚合酶γ导致的死亡。临床杂志。投资。2007;87:326–335. doi:10.1038/lipinvest.3700523。[内政部][PMC免费文章][公共医学][谷歌学者]
-
15Luoma P、Melberg A、Rinne JO等。帕金森综合征、更年期提前和线粒体DNA聚合酶γ突变:临床和分子遗传学研究。柳叶刀。2004;364:875–882. doi:10.1016/S0140-6736(04)16983-3。[内政部][公共医学][谷歌学者]
-
16Pagnameta AT、Taanman JW、Wilson CJ等。与线粒体DNA聚合酶γ突变相关的卵巢早衰显性遗传。哼。谴责。2006;21:2467–2473. doi:10.1093/humrep/del076。[内政部][公共医学][谷歌学者]
-
17Van Goethem G,Martin JJ,Dermaut B等。进展性外眼肌麻痹的复合杂合子患者中表现为感觉和共济失调性神经病变的隐性POLG突变。神经肌肉。迪索德。2003;13:133–142. doi:10.1016/s0960-8966(02)00216-x。[内政部][公共医学][谷歌学者]
-
18Naviaux RK,Nguyen KV。与阿尔卑斯综合征和线粒体DNA缺失相关的POLG突变。安。神经。2004;55:706–712. doi:10.1002/ana.20079。[内政部][公共医学][谷歌学者]
-
19Van Goethem G、Luoma P、Rantamaki M等。伴有共济失调但不涉及肌肉的神经退行性疾病中的POLG突变。神经病学。2004;63:1251–1257. doi:10.121/01.wnl.0000140494.58732.83。[内政部][公共医学][谷歌学者]
-
20Chan SSL、Longley MJ、Copeland WC。人类线粒体DNA聚合酶(POLG)中常见的A467T突变损害了催化效率以及与辅助亚单位的相互作用。生物学杂志。化学。2005;280:31341–31346. doi:10.1074/jbc。M506762200。[内政部][公共医学][谷歌学者]
-
21Naviaux RK、Nyhan WL、Barshop BA等。阿尔卑斯综合征患儿线粒体DNA聚合酶γ缺陷和线粒体DNA缺失。安。神经。1999;45:54–58. doi:10.1002/1531-8249(199901)45:1<54::aid-art10>3.0.co;2-b。[内政部][公共医学][谷歌学者]
-
22Chan-SSL、Longley MJ、Naviaux RK等。阿尔卑斯综合征患者非传感介导的衰变和选择性剪接导致的单等位基因POLG表达。DNA修复。2005;4:1381–1389. doi:10.1016/j.dnarep.2005.08.010。[内政部][公共医学][谷歌学者]
-
23Ferrari G、Lamantea E、Donati A等。与线粒体DNA聚合酶-γA突变相关的婴儿肝脑综合征。大脑。2005;128:723–731. doi:10.1093/brain/awh410。[内政部][公共医学][谷歌学者]
-
24Nguyen KV、Sharief F、Chan SSL等。阿尔卑斯综合征的分子诊断。《肝素杂志》。2006;45:108–116. doi:10.1016/j.jhep.2005.12.026。[内政部][公共医学][谷歌学者]
-
25Horvath R、Hudson G、Ferrari G等。与线粒体聚合酶γ基因突变相关的表型谱。大脑。2006;129:1674–1684. doi:10.1093/brain/awl088。[内政部][公共医学][谷歌学者]
-
26Hakonen AH、Heiskanen S、Juvonen V等。线粒体DNA聚合酶W748S突变:一种常见的欧洲起源的常染色体隐性共济失调原因。Am.J.Hum.遗传学。2005;77:430–441. doi:10.1086/444548。[内政部][PMC免费文章][公共医学][谷歌学者]
-
27Winterthun S、Ferrari G、He L等。线粒体聚合酶γ突变引起的常染色体隐性线粒体共济失调综合征。神经病学。2005;第64:1204–1208页。doi:10.1212/01.WNL.000156516.77696.5A。[内政部][公共医学][谷歌学者]
-
28Luoma PT,Luo N,Loscher WN,等。共济失调肌病综合征家族中人类线粒体DNA聚合酶间隔区突变导致的功能缺陷。嗯,分子遗传学。2005;1907年至1920年14时。doi:10.1093/hmg/ddi196。[内政部][公共医学][谷歌学者]
-
29Hudson G、Deschauer M、Taylor RW等。POLG1、C10ORF2和ANT1突变在伴有多个mtDNA缺失的散发性PEO中并不常见。神经病学。2006;66:1439–1441. doi:10.1212/01.wnl.0000210486.32196.24。[内政部][公共医学][谷歌学者]
-
30Chan SSL、Longley MJ、Copeland WC。线粒体疾病中E1143G多态性对DNA聚合酶γ中W748S突变的调节。嗯,分子遗传学。2006;15:3473–3483. doi:10.1093/hmg/ddl424。[内政部][PMC免费文章][公共医学][谷歌学者]
-
31宾夕法尼亚州罗普市,科普兰WC。人线粒体DNA聚合酶(DNA聚合物γ)的克隆和鉴定。基因组学。1996;36:449–458. doi:10.1006/geno.1996.0490。[内政部][公共医学][谷歌学者]
-
32Spelbrink JN、Toivonen JM、Hakkaart GA等。培养人类细胞中表达的人类线粒体DNA聚合酶POLG的体内功能分析。生物学杂志。化学。2000;275:24818–24828. doi:10.1074/jbc。M000559200。[内政部][公共医学][谷歌学者]
-
33Rovio AT、Marchington DR、Donat S等。线粒体DNA聚合酶(POLG)基因座突变与男性不育的关系。自然遗传学。2001;29:261–262. doi:10.1038/ng759。[内政部][公共医学][谷歌学者]
-
34Jensen M,Leffers H,Petersen JH,等。线粒体DNA聚合酶γ基因(POLG)在正常精子图和不明原因的低生育率患者中的频繁多态性。哼。谴责。2004;19:65–70. doi:10.1093/humrep/deh038。[内政部][公共医学][谷歌学者]
-
35Krausz C、Guarducci E、Becherini L等。POLG基因多态性在男性不育中的临床意义。临床杂志。内分泌代谢。2004;89:4292–4297. doi:10.1210/jc.2004-0008。[内政部][公共医学][谷歌学者]
-
36Aknin-Seifer IE、Touraine RL、Lejeune H等。线粒体DNA聚合酶γ(POLG)的CAG重复序列与男性不育相关吗?多中心法语学习。哼。谴责。2005;20:736–740. doi:10.1093/humrep/deh666。[内政部][公共医学][谷歌学者]
-
37Longley MJ、Clark S、Yu Wai Man C等。突变POLG2破坏DNA聚合酶γ亚单位并导致进行性眼肌麻痹。Am.J.Hum.遗传学。2006;78:1026–1034. doi:10.1086/504303。发现POLG2型polγ辅助亚单位也可引起adPEO。
-
38Spelbrink JN,Li FY,Tiranty V等。与编码Twinkle基因突变相关的人类线粒体DNA缺失,Twinkle是一种定位于线粒体中的噬菌体T7基因4-样蛋白。自然遗传学。2001;28:223–231. doi:10.1038/90058。线粒体DNA解旋酶基因的发现及其与PEO的关联。
-
39Garrido N,Griparic L,Jokitalo E等。人类线粒体类核的组成和动力学。分子生物学。单元格。2003;14:1583–1596. doi:10.1091/mbc。E02-07-0399。[内政部][PMC免费文章][公共医学][谷歌学者]
-
40Hudson G、Deschauer M、Busse K等。一种新的C10Orf2突变引起的感觉共济失调性神经病,可能存在种系嵌合体。神经病学。2005;64:371–373. doi:10.1212/01.WNL.000149767.51152.83。[内政部][公共医学][谷歌学者]
-
41Tynismaa H、Mjosund KP、Wanrooij S等。突变线粒体解旋酶Twinkle导致小鼠多个线粒体DNA缺失和迟发性线粒体疾病。程序。国家。阿卡德。科学。美国2005年;102:17687–17692。doi:10.1073/pnas.0505551102。[内政部][PMC免费文章][公共医学][谷歌学者]
-
42Kaukonen J、Zeviani M、Comi GP等。常染色体显性遗传进行性外眼肌麻痹中易导致mtDNA多重缺失的第三个基因座。Am.J.Hum.遗传学。1999;65:256–261. doi:10.1086/302445。[内政部][PMC免费文章][公共医学][谷歌学者]
-
43Koukonen J、Juselius JK、Tiranti V等。腺嘌呤核苷酸转运体1在线粒体DNA维护中的作用。科学。2000;289:782–785. doi:10.1126/science.289.5480.782。发现ANT1型是PEO所在地。
-
44Napoli L、Bordoni A、Zeviani M等。希腊adPEO家族中一种新的错义腺嘌呤核苷酸转运体-1基因突变。神经病学。2001;57:2295–2298. doi:10.1212/wnl.57.12.2295。[内政部][公共医学][谷歌学者]
-
45Agostino A、Valletta L、Chinnery PF等。ANT1、Twinkle和POLG1在散发性进行性外眼肌麻痹(PEO)神经病学中的突变。2003;60:1354–1356. doi:10.1212/01.wnl.0000056088.09408.3c。[内政部][公共医学][谷歌学者]
-
46Deschauer M、Hudson G、Muller T等。常染色体显性进展性外眼肌麻痹中一种新的ANT1基因突变,可能存在种系嵌合体。神经肌肉。迪索德。2005;15:311–315. doi:10.1016/j.nmd.2004.12.004。[内政部][公共医学][谷歌学者]
-
47Fontanesi F、Palmieri L、Scarcia P等。AAC2突变相当于人类adPEO相关ANT1突变,导致酿酒酵母中氧化磷酸化缺陷,并影响线粒体DNA稳定性。嗯,分子遗传学。2004;13:923–934. doi:10.1093/hmg/ddh108。[内政部][公共医学][谷歌学者]
-
48Nishino I,Spinazzola A,Hirano M.人类线粒体疾病MNGIE中的胸苷磷酸化酶基因突变。科学。1999;283:689–692. doi:10.126/science.283.5402.689。胸苷磷酸化酶是发现的第一个与线粒体DNA突变疾病相关的蛋白质,MNGIE。
-
49Hirano M、Lagier-Tourenne C、Valentino ML等。胸苷磷酸化酶突变导致线粒体DNA不稳定。基因。2005;354:152–156. doi:10.1016/j.gene.2005.04.041。[内政部][公共医学][谷歌学者]
-
50Hirano M,Nishigaki Y,Marti R.线粒体神经胃肠道脑肌病(MNGIE):一种由两个基因组组成的疾病。神经学家。2004;10:8–17. doi:10.1097/01.nrl.000106919.06469.04。[内政部][公共医学][谷歌学者]
-
51Nishino I、Spinazzola A、Papadimitriou A等。线粒体神经胃肠道脑肌病:一种由胸苷磷酸化酶突变引起的常染色体隐性疾病。安。神经。2000;47:792–800.[公共医学][谷歌学者]
-
52Spinazzola A、Marti R、Nishino I等。胸腺嘧啶磷酸化酶缺陷导致的胸腺嘧啶代谢改变。生物学杂志。化学。2001;3:3. doi:10.1074/jbc。M111028200。[内政部][公共医学][谷歌学者]
-
53Marti R,Nishigaki Y,Hirano M。胸苷磷酸化酶缺乏症患者血浆脱氧尿苷升高。生物化学。生物物理学。Res.Commun公司。2003;303:14–18. doi:10.1016/s0006-291x(03)00294-8。[内政部][公共医学][谷歌学者]
-
54Song S,Wheeler LJ,Mathews CK。脱氧核糖核苷酸库失衡刺激HeLa细胞线粒体DNA缺失。生物学杂志。化学。2003;278:43893–43896. doi:10.1074/jbc。C300401200。[内政部][公共医学][谷歌学者]
-
55Nishigaki Y、Marti R、Copeland WC等。胸苷磷酸化酶缺乏症患者的位点特异性线粒体DNA点突变。临床杂志。投资。2003;111:1913–1921. doi:10.1172/JCI17828。[内政部][PMC免费文章][公共医学][谷歌学者]
-
56Longley MJ、Nguyen D、Kunkel TA等。人类DNA聚合酶γ的保真度(有无外核裂解校对和p55辅助亚单位)。生物学杂志。化学。2001;276:38555–38562. doi:10.1074/jbc。M105230200。[内政部][公共医学][谷歌学者]
-
57Lim SE,科普兰WC。人类DNA聚合酶γ对抗病毒脱氧核苷酸的差异掺入和去除。生物学杂志。化学。2001;276:23616–23623. doi:10.1074/jbc。M10114200。[内政部][公共医学][谷歌学者]
-
58Spinazzola A、Marti R、Nishino I等。胸腺嘧啶磷酸化酶缺陷导致的胸腺嘧啶代谢改变。生物学杂志。化学。2002;277:4128–4133. doi:10.1074/jbc。M111028200。[内政部][公共医学][谷歌学者]
-
59Hirano M、Marti R、Casali C等。异基因干细胞移植纠正MNGIE中的生化紊乱。神经病学。2006;67:1458–1460. doi:10.121/01.wnl.0000240853.97716.24。[内政部][PMC免费文章][公共医学][谷歌学者]
-
60Lara MC、Weiss B、Illa I等。短暂输注血小板可降低MNGIE的核苷过载。神经病学。2006;67:1461–1463. doi:10.1212/01.wnl.0000239824.95411.52。[内政部][公共医学][谷歌学者]
-
61Saada A、Shaag A、Mandel H等。线粒体DNA缺失性肌病中的线粒体胸苷激酶突变。自然遗传学。2001;29:342–344. doi:10.1038/ng751。第一份报告描述TK2型线粒体DNA缺失性肌病的位点。
-
62Mancuso M、Salviati L、Sacconi S等。线粒体DNA缺失:胸腺嘧啶激酶基因突变与肌病和SMA。神经病学。2002;59:1197–1202。doi:10.1212/01.wnl.0000028689.93049.9a。[内政部][公共医学][谷歌学者]
-
63Mancuso M、Filosto M、Bonilla E等。与线粒体DNA缺失和TK2基因纯合突变(T77M)相关的儿童线粒体肌病。架构(architecture)。神经醇。2003;60:1007–1009. doi:10.1001/archneur.60.7.1007。[内政部][公共医学][谷歌学者]
-
64Carrozzo R,Bornstein B,Lucioli S等。16例mtDNA缺失患者的突变分析。嗯,变种人。2003;21:453–454. doi:10.1002/humu.9135。[内政部][公共医学][谷歌学者]
-
65Wang L,Limongelli A,Vila MR,等。两名脱氧鸟苷激酶和胸苷激酶2基因新突变患者线粒体DNA缺失综合征的分子观察。分子遗传学。代谢产物。2005;84:75–82. doi:10.1016/j.ymgme.2004.09.005。[内政部][公共医学][谷歌学者]
-
66.Galbiati S、Bordoni A、Papadimitriou D等。与线粒体DNA缺失相关的TK2基因新突变。儿科。神经醇。2006;34:177–185. doi:10.1016/j.pediatreneul.2005.07.013。[内政部][公共医学][谷歌学者]
-
67Alberio S,Mineri R,Tiranti V等人,线粒体DNA的消耗:综合征和基因。线粒体。2007;7:6–12. doi:10.1016/j.mito.2006.11.010。[内政部][公共医学][谷歌学者]
-
68Saada A,Shaag A,Elpeleg O。线粒体DNA缺失性肌病:线粒体胸苷激酶(TK2)缺乏症组织特异性的阐明。分子遗传学。代谢产物。2003;79:1–5. doi:10.1016/s1096-7192(03)00063-5。[内政部][公共医学][谷歌学者]
-
69Mandel H、Szargel R、Labay V等。脱氧鸟苷激酶基因在肝脑线粒体DNA缺失的个体中发生突变。自然遗传学。2001;29:337–341. doi:10.1038/ng746。首次报道脱氧鸟苷激酶基因参与线粒体DNA缺失。
-
70Salviati L、Sacconi S、Mancuso M等。线粒体DNA缺失和dGK基因突变。安。神经。2002;52:311–317. doi:10.1002/ana.10284。[内政部][公共医学][谷歌学者]
-
71Wang L,Eriksson S.线粒体脱氧鸟苷激酶突变与线粒体DNA缺失综合征。FEBS信函。2003;554:319–322. doi:10.1016/s0014-5793(03)01181-5。[内政部][公共医学][谷歌学者]
-
72Freisinger P、Futterer N、Lankes E等。脱氧鸟苷激酶(DGUOK)突变引起的肝脑线粒体DNA缺失综合征。架构(architecture)。神经醇。2006;63:1129–1134. doi:10.1001/archneur.63.8.1129。[内政部][公共医学][谷歌学者]
-
73Kelley RI、Robinson D、Puffenberger EG等。阿米什致命小头畸形:一种新的代谢紊乱,伴有严重先天性小头畸形和2-酮戊二酸尿症。美国医学遗传学杂志。2002;112:318–326. doi:10.1002/ajmg.10529。[内政部][公共医学][谷歌学者]
-
74Rosenberg MJ、Agarwala R、Bouffard G等。突变脱氧核苷酸携带者与先天性小头畸形相关。自然遗传学。2002;32:175–179. doi:10.1038/ng948。[内政部][公共医学][谷歌学者]
-
75Lindhurst MJ、Fiermonte G、Song S等。Slc25a19基因敲除导致线粒体焦磷酸硫胺耗竭、胚胎致死、中枢神经系统畸形和贫血。程序。国家。阿卡德。科学。美国2006年;103:15927–15932。doi:10.1073/pnas.0607661103。[内政部][PMC免费文章][公共医学][谷歌学者]
-
76Marobbio CM、Vozza A、Harding M等。焦磷酸硫胺酵母线粒体转运体的鉴定和重组。EMBO J.2002;21:5653–5661. doi:10.1093/emboj/cdf583。[内政部][PMC免费文章][公共医学][谷歌学者]
-
77Elpeleg O、Miller C、Hershkovitz E等。ADP-形成琥珀酰-CoA合成酶活性缺乏与脑肌病和线粒体DNA缺失相关。Am.J.Hum.遗传学。2005;76:1081–1086。doi:10.1086/430843。柠檬酸循环基因也可导致线粒体DNA缺失综合征。
-
78Ostergaard E、Hansen FJ、Sorensen N等。甲基丙二酸升高的线粒体脑肌病是由SUCLA2突变引起的。大脑。2007;130:853–861. doi:10.1093/brain/awl383。[内政部][公共医学][谷歌学者]
-
79Carrozzo R、Dionisi-Vici C、Steuerwald U等。SUCLA2突变与轻度甲基丙二酸尿、Leigh样脑肌病、肌张力障碍和耳聋相关。大脑。2007;130:862–874. doi:10.1093/brain/awl389。[内政部][公共医学][谷歌学者]
-
80Kowluru A,Tannous M,Chen总部。胰岛β细胞核苷二磷酸激酶线粒体亚型的定位和特征:其与线粒体琥珀酰辅酶A合成酶复合的证据。架构(architecture)。生物化学。生物物理学。2002;398:160–169. doi:10.1006/abbi.2001.2710。[内政部][公共医学][谷歌学者]
-
81Spinazzola A、Viscomi C、Fernandez-Vizarra E等。MPV17编码一种内线粒体膜蛋白,在婴儿肝脏线粒体DNA缺失时发生突变。自然遗传学。2006;38:570–575. doi:10.1038/ng1765。利用综合基因组学鉴定新基因,MPV17型与线粒体DNA缺失综合征和肝病有关。
-
82Calvo S,Jain M,Xie X,等。通过整合基因组学系统鉴定人类线粒体疾病基因。自然遗传学。2006;38:576–582. doi:10.1038/ng1776。[内政部][公共医学][谷歌学者]
-
83Karadimas CL、Vu TH、Holve SA等。纳瓦霍神经性肝病是由MPV17基因突变引起的。Am.J.Hum.遗传学。2006;79:544–548. doi:10.1086/506913。[内政部][PMC免费文章][公共医学][谷歌学者]
-
84Meyer-zum Gottesberge AM,Reuter A,Weiher H.肾小球硬化小鼠模型Mpv17中类似Alport综合征的内耳缺损。欧洲体系结构。耳鼻咽喉科。1996;253:470–474. doi:10.1007/BF00179952。[内政部][公共医学][谷歌学者]
-
85Bourdon A,Minai L,Serre V等。编码p52控制的核糖核苷酸还原酶(p53R2)的RRM2B突变会导致线粒体DNA严重缺失。自然遗传学。2007;39:776–780. doi:10.1038/ng2040。[内政部][公共医学][谷歌学者]

