摘要
蛋白精氨酸甲基转移酶PRMT5与人SWI/SNF复合物和甲基化组蛋白H3R8和H4R3相互作用。为了阐明PRMT5在人类癌症中的作用,我们分析了正常人B淋巴细胞、一组淋巴癌细胞系以及套细胞淋巴瘤(MCL)临床样本中PRMT5的表达。我们发现,尽管PRMT5的转录速率和信使RNA的稳定性较低,但在所有癌细胞中,包括所检测的临床样本中,PRMT5蛋白水平均升高。值得注意的是,多聚体分析显示PRMT5项目与正常B细胞相比,Mino和JeKo MCL细胞中mRNA的翻译效率更高,miR-92b和miR-96表达的减少增加了PRMT5的翻译。因此,H3R8和H4R3的整体甲基化增加,并伴有淋巴癌细胞中肿瘤发生性抑制因子7(ST7)的抑制。此外,下调PRMT5表达可减少转化的JeKo和Raji细胞的增殖。因此,我们的研究表明,PRMT5的异常表达导致染色质的表观遗传修饰发生改变,进而影响抗癌基因的转录性能和转化淋巴细胞的生长。
关键词:H3R8和H4R3甲基化,套细胞淋巴瘤,miR-92b和miR-96,PRMT5,ST7标准
介绍
人类恶性肿瘤是由于调节细胞生长和增殖的基因的异常表达引起的,了解导致基因表达改变的分子机制对于设计专门针对受影响的中央调节网络的疗法很重要。最近的研究表明,染色质修饰酶可以激活或抑制基因表达,在癌症病因学中起着关键作用(圣罗萨和卡尔达斯,2005年;埃斯特勒,2006). 染色质可以通过各种机制改变,包括组蛋白和/或DNA的共价修饰和核小体内组蛋白与DNA接触的非共价扰动。这些染色质改变的最终结果是调节阻遏蛋白和激活蛋白的可及性(Sif,2004年;Martin和Zhang,2005年). 一些报告表明,染色质修饰酶之间的相互作用和串扰对于有效调节基因表达是必要的,而影响染色质重塑器活性或靶向性的变化可能会引发癌症(菲施勒等, 2003;哈克等, 2004).
组蛋白修饰如乙酰化、甲基化和泛素化已成为基因表达控制中的重要标志,它们可以协同或拮抗作用来指定转录性能(菲施勒等, 2003). 组蛋白赖氨酸和精氨酸残基都可以分别被SET蛋白和蛋白精氨酸甲基转移酶(PRMT)甲基化,不同小组的研究表明,甲基化可以根据被修饰的类型和残基抑制或诱导转录(贝德福德和理查德,2005年;Martin和Zhang,2005年). 例如,H3K4的三甲基化与活性转录相关,并与H3和H4乙酰化协同作用以激活Hoxc8型表达式(米尔恩等2002年). 相反,H3K9和/或H3K27的三甲基化诱导基因沉默,似乎被排除在超双胸携带三甲基化H3K4的启动子区域(曹等2002年;霍尔等2002年;帕普和穆勒,2006年).
PRMTs在进化上是保守的,PRMT5是催化ω-N个克-单甲基化和ω-N个克,N个G′-对称二甲基化(贝德福德和理查德,2005年). PRMT5与许多多亚单位复合物相互作用,参与多种细胞过程,包括RNA处理、信号转导、转录调控和生殖细胞发育(波拉克等, 1999;弗里森等, 2001;法布里齐奥等2002年;朋友等, 2003,2004;安切林等, 2006). 有趣的是,PRMT5可以以甲基化酶依赖或非依赖的方式影响转录。作为雄激素受体辅因子复合物的一种成分,PRMT5独立于其甲基转移酶活性,积极调节雄激素受体驱动的转录(Hosohata公司等, 2003); 然而,当PRMT5与hSWI/SNF复合物相关时,其染色质修饰活性对组蛋白H3和H4甲基化以及转录调控非常重要(朋友等, 2004;Dacwag公司等, 2007). 此外,PRMT5的甲基化也可能对其底物产生积极或消极的影响。例如,PRMT5介导的SmD1和SmD3的甲基化对它们并入参与RNA剪接的snRNP至关重要,而NURD MBD2亚单位的甲基化降低了其与甲基化DNA结合和抑制转录的能力(弗里森等, 2001;古岑内克等, 2006;Tan和Nakielny,2006年).
最近的研究表明,PRMT5既可以通过修饰组蛋白直接调节基因表达,也可以通过改变各种转录因子的活性间接调节基因表达(法布里齐奥等2002年;快克等, 2003;朋友等, 2003,2004;盖森内克等, 2006;Tan和Nakielny,2006年). PRMT5参与转录调控的直接证据来自于明确证明PRMT5与染色质重塑复合物相互作用的研究,如BRG1/BRM-based hSWI/SNF复合物和MBD2-based NURD复合物(朋友等, 2003,2004;盖森内克等, 2006). 作为这些染色质重塑物的一部分,PRMT5能够甲基化组蛋白H3和H4,并抑制肿瘤抑制基因的转录,如肿瘤发生性抑制因子7(ST7标准)和NM23-H1型和细胞周期调节器,如循环素E1,第14页农业研究基金和第16页INK4a公司.
为了评估PRMT5在人类淋巴癌中的作用,我们分析了其在一组转化淋巴细胞中的表达,包括套细胞淋巴瘤(MCL)细胞系和MCL患者样本。MCL是非霍奇金B细胞淋巴瘤的一种亚型,其特征是存在t(11;14)(q13;q32)染色体易位。MCL患者预后极差,生存率为3-4年。由于缺乏有效的治疗手段,MCL是无法治愈的,新的治疗靶点的确定仍然是MCL研究的重点(Witzig,2005年). 以患者衍生MCL细胞系为模型系统,我们检测了PRMT5水平和组蛋白H3R8和H4R3的整体甲基化,组蛋白H3和H4R优先被PRMT5对称甲基化。我们发现PRMT5在多种淋巴癌细胞系中高度表达,包括MCL临床样本,因此H3R8和H4R3的全局对称甲基化在转化的淋巴细胞系和MCL临床样本中也发生了改变。我们还表明,PRMT5蛋白表达的增加在翻译水平上受到调节,并与转录沉默ST7标准此外,敲低PRMT5表达会干扰转化的B细胞的生长。因此,我们的工作将PRMT5确定为一个关键染色质调节器,其异常表达影响H3R8和H4R3的整体甲基化,有助于ST7标准抑癌基因沉默并与淋巴腺病相关。
结果
PRMT5在淋巴癌细胞系中过度表达,其水平与组蛋白H3R8和H4R3对称甲基化增加相关
我们之前已经证明,PRMT5可以与BRG1和基于BRM的hSWI/SNF相互作用,并且PRMT5在永生化但未转化的细胞系中的过度表达可以诱导过度增殖和细胞转化(朋友等, 2004). 后一结果促使我们研究了PRMT5在人类淋巴源性恶性肿瘤中的作用,这些恶性肿瘤的正常对照容易获得。正常CD19的核和细胞溶质提取物+B淋巴细胞、患者衍生淋巴瘤(Burkitt淋巴瘤:Daudi、Jijoye、BL30、P3HRI和Raji;MCL:Mino、JeKo和SP53),在体外用Western blotting分析EBV转化淋巴瘤(LCL-147)和白血病细胞系(EOL1、NB4、Mv411)(图1A). 转化淋巴细胞的核和细胞溶质部分的PRMT5蛋白水平均升高。由于PRMT5与BRG1和BRM染色质重塑复合物相关,hSWI/SNF组分的突变与包括白血病和淋巴瘤在内的各种人类癌症有关,我们分析了正常和转化淋巴细胞中BRG1、BRM和BAF57的水平。hSWI/SNF亚单位在所有样本中均有表达,但BRM表达降低的Burkitt淋巴瘤P3HRI细胞系除外。由于PRMT5存在于细胞核和细胞溶质部分,我们测量了α-管蛋白的表达,发现它只在细胞溶质中表达,表明细胞核中PRMT5的存在不是由于细胞溶质污染。
图1。
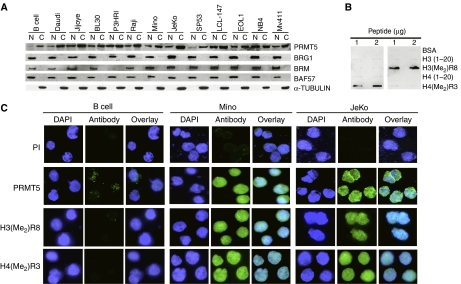
PRMT5在淋巴瘤和白血病细胞系中过表达。(A类)使用正常CD19的核(N)和细胞溶质(C)提取物通过Western blotting评估PRMT5和hSWI/SNF亚单位的表达+B细胞或所示转化细胞系(20μg)。为了区分N和C组分,我们测量了α-TUBULIN的表达。(B类)抗-H3(Me2)R8和抗H4(Me2)R3抗体不交叉反应,具有高度特异性。将BSA或指示肽的部分(1和2μg)标记在硝化纤维素膜上,并使用抗H4(Me)检测2)R3或抗H3(Me2)R8(C类)PRMT5、H4(Me)的免疫荧光2)R3和H3(Me2)正常B细胞、Mino细胞和JeKo细胞中的R8。将正常B细胞和MCL细胞固定并与免疫前或免疫性抗PRMT5、抗H3(Me2)R8和H4(Me2)R3抗体。FITC标记的山羊抗兔抗体用于检测PRMT5和修饰组蛋白,DAPI用于细胞核染色。照片是放大100倍拍摄的。
为了进一步证实Western blot数据,我们用免疫前或免疫性抗PRMT5抗体对正常B细胞和患者来源的Mino和JeKo MCL细胞进行染色(图1C). 仅当使用免疫性抗PRMT5抗体时检测到一个信号,该抗体在Mino和JeKo MCL细胞系中显著升高,表明转化淋巴细胞中PRMT5蛋白水平升高。我们之前已经证明PRMT5可以甲基化组蛋白H3R8和H4R3在体外,并且我们已经将PRMT5募集与H3R8的对称甲基化联系起来体内(朋友等, 2004). 因此,我们使用能够特异识别对称甲基化H3R8或H4R3的抗体,分析了正常B细胞和MCL细胞中H3R4和H4R8的整体甲基化(图1B、和补充图1A和B). 使用全细胞提取物对两种抗体的进一步表征表明,抗H3(Me2)R8抗体具有高度特异性,而抗H4(Me2)除了对称甲基化的H4R3外,R3抗体还与其他四种多肽弱交叉反应(补充图1C和E). 当抗H3(Me2)R8和抗H4(Me2)R3抗体用于免疫荧光研究,但在正常B淋巴细胞中检测不到这些表观遗传标记;然而,Mino和JeKo MCL细胞系表现出高水平的对称甲基化H3R8和H4R3(图1C). 用抗H3(Me2)R8和抗H4(Me2)R3抗体(补充图1D和E). 总之,这些研究表明,PRMT5的表达增加导致H3R8和H4R3的对称甲基化,并与MCL病理学相关。
患者源性Mino和JeKo MCL细胞系中PRMT5 mRNA的表达和稳定性发生改变
发现PRMT5蛋白在各种转化的淋巴细胞系中的表达增强,我们想确定这是否是由于PRMT5项目信使核糖核酸表达(图2A). 实时RT-PCR显示,尽管癌细胞中PRMT5蛋白水平升高PRMT5项目在转化的淋巴细胞中,mRNA降低了2到5倍(P(P)<10−4)这表明还有其他机制参与PRMT5蛋白表达的上调。了解减少的根本原因PRMT5项目mRNA在转化淋巴细胞中的表达,我们检测了PRMT5项目正常B细胞以及患者衍生Mino和JeKo MCL细胞系的转录和mRNA稳定性(图2B和C). 与实时RT-PCR结果一致,核试运行分析表明PRMT5项目米诺人的mRNA转录分别低2.9倍和2.4倍(P(P)=0.028)和JeKo(P(P)=0.011)MCL细胞系,表明PRMT5项目mRNA转录在正常B淋巴细胞中更有效。测量PRMT5项目用DRB处理mRNA稳定性、正常B淋巴细胞以及Mino和JeKo MCL细胞系以抑制mRNA合成PRMT5项目通过实时RT-PCR测量不同时间的mRNA水平。18秒rRNA被用作内部对照以正常化PRMT5项目这些实验中的mRNA水平,因为低剂量和短时间的DRB治疗不会影响RNA聚合酶I转录。我们发现,与正常B细胞相比(吨1/2=226分钟),半衰期PRMT5项目Mino的信使核糖核酸减少了2.7倍(吨1/2=84分钟),JeKo为2.3倍(吨1/2=96分钟)MCL细胞系(P(P)=0.009). 综上所述,这些发现以及观察到的PRMT5项目mRNA转录表明,观察到的PRMT5稳态水平的增加可能是两种增加的结果PRMT5项目mRNA翻译和/或增强转化MCL细胞系中PRMT5蛋白的稳定性。
图2。
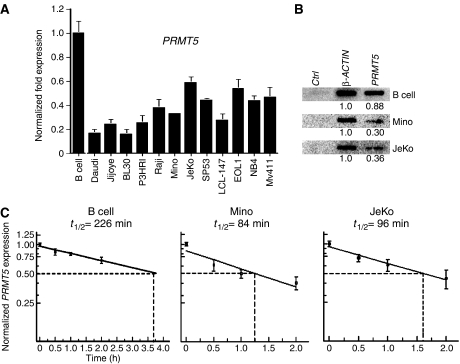
的表达式PRMT5项目在不同级别进行监管。(A类)PRMT5项目用实时RT-PCR检测正常和转化B细胞的mRNA表达。条形图显示了PRMT5项目mRNA在不同细胞系相对于正常B细胞中的表达GAPDH公司作为内部控制。RT-PCR分析分三次进行,共三次,图表显示平均值±标准差(B类)PRMT5项目MCL细胞系中的转录发生改变。按照材料和方法所述进行核试运行分析。控件(Ctrl)Pvu公司II–Pvu公司pBluescript KS(+)和β的II DNA片段-肌动蛋白和PRMT5项目cDNA PCR片段固定在Hybond XL膜上,并用从指示细胞分离的放射性标记RNA进行检测。信号被量化PRMT5项目与β有关-肌动蛋白. (C类)PRMT5项目mRNA在B细胞中更稳定。将正常和转化的B细胞用DRB处理指定时间,并通过实时RT-PCR分离和定量总RNA。PRMT5项目mRNA表达标准化使用18秒作为内部控制。计算半衰期(吨1/2)第页,共页PRMT5项目mRNA,每个图中的数据点是由四个不同的实验产生的,实验进行了三次。
miR-92b和miR-96的异常表达与PRMT5翻译增强有关
分析PRMT5项目mRNA翻译,我们使用正常B淋巴细胞和MCL细胞系的细胞裂解物在放线菌酮存在下进行蔗糖梯度沉淀实验(图3A). 收集梯度部分,提取总RNA并用于测量PRMT5项目实时RT-PCR检测mRNA。β的量-肌动蛋白将每个级分中的mRNA用作内部对照,以使PRMT5项目mRNA存在于每一部分中。蔗糖梯度分析表明,尽管PRMT5项目在正常和转化的B细胞的多核糖体组分11~18中检测到mRNA,仅占总数的24%PRMT5项目正常B淋巴细胞的mRNA为多核糖体。然而PRMT5项目与多核糖体相关的mRNA在米诺中富集了2.2倍和2.6倍(P(P)<10−4)和JeKo(P(P)=0.0009)个单元格。这种多核糖体图谱的变化PRMT5项目mRNA和Western blot结果表明PRMT5项目mRNA翻译在转化的Mino和JeKo MCL细胞系中增强。
图3。
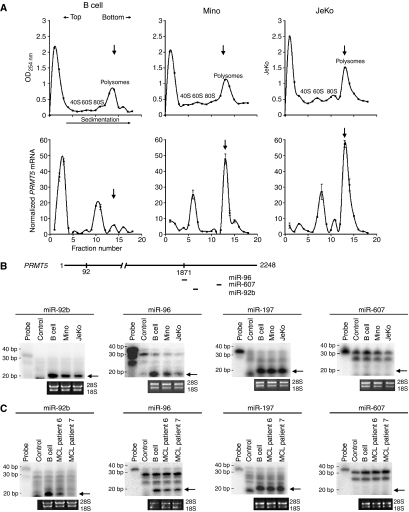
PRMT5项目mRNA在MCL细胞系中的翻译效率更高。(A类)正常和转化B细胞的多核糖体图谱。通过15–40%蔗糖梯度沉淀对全细胞裂解液进行分级,并通过测量每个部分在254 nm处的吸光度来确定多核糖体轮廓(上面板)。显示了代表40S、60S、80S和多核糖体的部分。从每个部分分离的RNA用于测量PRMT5项目实时RT-PCR检测mRNA和β-肌动蛋白被用作内部控制来规范PRMT5项目各部分的mRNA水平(下面板)。金额PRMT5项目每个片段中存在的mRNA相对于含有最低拷贝数的片段进行报告PRMT5项目mRNA。每个图中的数据点表示三次RT-PCR反应的平均值±s.d。垂直箭头表示峰值多核糖体部分。(B、 C类)的差异表达PRMT5项目-正常和转化B细胞中的特异性miRNAs。的示意图PRMT5项目描绘3′UTR内潜在miRNA-结合位点位置的mRNA(B,上面板)。使用miR-92b、miR-96、miR-197或miR-607探针对从指示细胞中分离的20μg(B)或5μg(C)总RNA进行RPA。探针代表每个反应中使用的标记探针总量的1/10,对照组显示探针在酵母tRNA存在下消化。包括溴化乙锭凝胶,以显示相等的负载。箭头显示成熟miRNAs的位置。
阐明增加翻译的机制PRMT5项目,我们推断可能有PRMT5项目-参与调节其翻译的特异性miRNA。我们搜索了Wellcome Trust Sanger Institute miRNA注册库(http://microrna.sanger.ac.uk)在细胞的3′非翻译(UTR)区域存在潜在的miRNA结合位点PRMT5项目.我们分析了11个电位的表达PRMT5项目使用RNase保护试验靶向正常和转化B细胞中的miRNA(图3B和C和数据未显示)。在所有测试的miRNAs中,仅检测到miR-92b和miR-96的表达;然而,它们在Mino中的表达降低了2到2.5倍(miR-92bP(P)=0.009,miR-96P(P)=0.012)和JeKo(miR-92bP(P)=0.01,miR-96P(P)=0.005)MCL细胞系(图3B). 当我们分析一种不相关的对照miRNA miR-197的水平时,我们发现其在正常和转化的B细胞中的表达没有改变。更重要的是,我们还发现在MCL临床样本6和7中miR-92b和miR-96的表达降低,这表明这两种miRNAs的异常表达是MCL的标志(图3C).
miR-92b和miR-96的再表达抑制PRMT5翻译体内,其结合位点对翻译调控至关重要
进一步评估miR-92b和miR-96对PRMT5翻译调控的影响体内,我们分别用野生型或突变型miR-92b和miR-96电穿孔JeKo细胞,并检查PRMT5蛋白的水平(图4A). 在存在野生型miR-92b的情况下,PRMT5蛋白表达降低80%,而miR-96导致蛋白表达降低50%。类似地,当我们在Raji细胞中进行相同的实验时,PRMT5蛋白水平降低了60-85%,突显了miR-92b和miR-96表达在PRMT5翻译调控中的重要性(图4A). 验证PRMT5表达减少是否是翻译抑制的结果,但不是PRMT5项目mRNA降解,我们测量PRMT5项目模拟转染细胞以及野生型和突变型miR-92b或miR-96转染细胞的mRNA稳态水平(补充图2A和B). 正如预期的那样,在JeKo和Raji细胞系中存在外源性转染的miR-92b或miR-96时,PRMT5 mRNA水平没有显著改变。正如我们之前确定的那样ST7标准作为PRMT5靶基因,我们分析了其在miR-92b或miR-96电穿孔Raji细胞中的mRNA水平(补充图2C). 在miR-92b存在的情况下,ST7标准mRNA水平下降了2.2倍(P(P)=0.001),而miR-96的电迁移导致1.9倍ST7标准减压(P(P)=0.005). 综上所述,这些结果表明PRMT5表达减少对ST7标准转录。
图4。
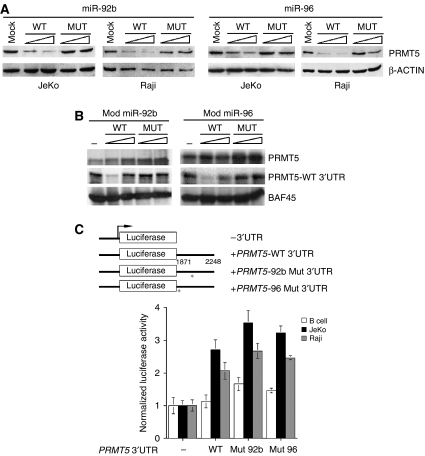
在转化的B细胞中,miR-92b和miR-96下调了PRMT5的表达。(A类)分别用2.5和5.0μg野生型或突变型miR-92b和miR-96双链RNA电穿孔JeKo和Raji细胞,并用Western blotting分析20μg RIPA提取物。(B类)修饰的miR-92b和miR-96对PRMT5表达的影响在体外.体外在没有或存在更多修饰野生型或突变型miR-92b或miR-96的情况下进行翻译,使用0.25μgPRMT5项目mRNA不含3′UTR(PRMT5)和野生型3′UTR-(PRMT5-WT 3′UTR.)。BAF45型mRNA作为对照。(C类)普通B(25×106)或改造JeKo和Raji(5×106)在pRL-TK存在下,用pCMV-LUC、融合到野生型PRMT5 3′UTR的pCMV-LUC或融合到突变的miR-92b或miR-96结合位点PRMT5 3'UTR构建物的pCMV LUC电穿孔细胞,并使用双荧光素酶报告分析测定荧光素素酶表达。荧光素酶活性是相对于每个细胞系的pCMV-LUC来表示的,并且已经使用Renilla荧光素酶进行了标准化。
接下来,我们重建了miR-92b和miR-96介导的翻译抑制体外PRMT5最近的研究表明,miRNAs的翻译基因沉默在体外需要7-甲基G帽和聚(a)尾(王等, 2006). 因此,我们在体外7-甲基G-封端和聚(A)尾的翻译实验PRMT5项目带有和不带有野生型3′UTR的mRNA。野生型和突变型miR-96均未能抑制PRMT5翻译在体外,表明兔网织红细胞裂解物中还缺少其他成分,这些成分可能会稳定miR-96与其靶位点的结合(数据未显示)。因为miR-96退火到PRMT5项目3′UTR主要通过其种子序列(+2到+8),我们假设PRMT5项目mRNA:miR-96杂合分子在30°C(ΔG=−13.76 kcal/mol,T型m=22°C)。为了弥补稳定性的不足,我们改变了miR-96中六个连续的核糖核苷酸(+9到+14)(ΔG=-27.5 kcal/mol,Tm=44°C),并测试了其抑制PRMT5翻译的能力在体外(图4B). 改良野生型,但不是突变型miR-96,其中种子序列(+2到+8)发生了突变,当PRMT5项目使用野生型3′UTR mRNA。的翻译PRMT5项目mRNA无3′UTRBAF45型mRNA未受影响。在这些试验中使用改良野生型和突变型miR-92b时,也观察到类似的结果在体外翻译实验(图4B).
根据这些发现,miR-92b或miR-96的重新表达降低了PRMT5的表达体内,我们想确定miR-92b和miR-96的结合位点是否对翻译调控至关重要(图4C). 野生型和突变型PRMT5项目3′UTR在CMV驱动荧光素酶报告子下游亚克隆,并电穿孔至正常B淋巴细胞或转化的JeKo和Raji细胞系。存在野生型PRMT5项目B细胞中3′UTR、荧光素酶的表达没有变化,而JeKo和Raji细胞中的表达增强了2.7-(P(P)=0.004)和2倍(P(P)=0.026),这表明miR-92b和miR-96在转化淋巴细胞中的表达降低可增强荧光素酶的表达。当miR-92b或miR-96种子序列结合位点发生突变时,萤光素酶的表达增加1.5-(P(P)=0.035)至1.7倍(P(P)=0.001)在B细胞中,3.2-(P(P)=0.006)至3.5倍(P(P)=0.003)在JeKo细胞中,以及2.5-(P(P)=0.037)至2.7倍(P(P)=0.008)。这些结果表明,miR-92b和miR-96有助于正确调节PRMT5翻译。
PRMT5靶基因ST7在淋巴癌细胞系中被抑制
我们之前已经确定ST7标准作为PRMT5的直接靶基因,并使用标记的PRMT5细胞系证明了PRMT5招募到ST7标准启动子诱导组蛋白H3R8对称甲基化,并与ST7标准转录抑制(朋友等, 2004). 由于一系列转化淋巴细胞系中PRMT5蛋白水平异常高,我们研究了PRMT5水平升高是否会影响ST7标准在这些转化细胞系中表达。实时RT-PCR分析表明ST7标准癌细胞株中mRNA表达减少了2到12倍(P(P)<10−4) (图5A). 然而,PRMT5蛋白水平与ST7标准mRNA抑制。值得注意的是,当用Western blotting检测ST7蛋白水平时,所有转化的淋巴细胞株都缺乏ST7表达,这表明PRMT5和ST7蛋白之间存在反向关系(图5B). 为了进一步证实这些结果,我们在正常B细胞以及Mino和JeKo MCL细胞系中通过免疫荧光检测到ST7蛋白。与Western blot数据一致,ST7在正常B细胞中检测到,但在转化的MCL细胞系中未检测到(图5C). 值得注意的是,当使用核或细胞溶质提取物时,未检测到ST7的表达(数据未显示);然而,当使用RIPA缓冲液制备全细胞裂解物时,ST7是容易检测到的。这些结果和正常B淋巴细胞的免疫荧光染色模式表明,ST7蛋白是不溶性的,可能是膜结合的。
图5。
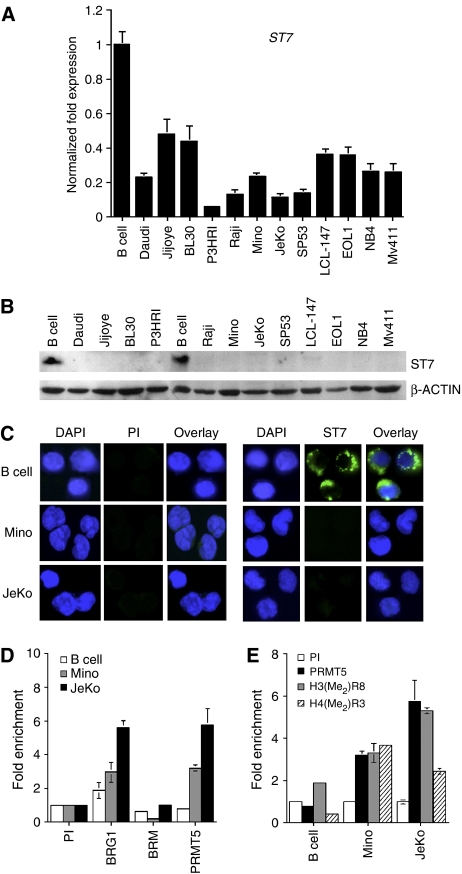
ST7标准在转化的淋巴细胞系中沉默。(A类)ST7标准实时RT-PCR检测到淋巴癌细胞系中的转录受到抑制。这个实验重复了三次,共三次。(B类)使用20μg来自正常B细胞或所示转化淋巴细胞系的RIPA提取物,通过Western blotting分析ST7蛋白表达。β-肌动蛋白检测水平以确保负载相等。(C类)固定的正常或转化的B细胞与免疫前或免疫性抗ST7抗体孵育。用山羊FITC-标记的抗兔抗体显示ST7蛋白,而细胞核用DAPI染色。照片是放大100倍拍摄的。(D类,E类)使用免疫前(PI)或指示的免疫抗体对正常或转化B细胞的交联染色质进行ChIP分析,并使用实时PCR扩增保留的DNAST7标准-特异性引物和探针。相对于PI样品计算每个抗体的折叠富集。每个ChIP实验重复两次,共三次。
的表达式ST7标准mRNA在所有被检测的转化淋巴细胞系中被抑制,表明ST7标准这些癌细胞系中的启动子活性发生改变。验证PRMT5是否直接参与诱导ST7标准抑制,我们使用抗PRMT5特异性抗体进行染色质免疫沉淀(ChIP)实验(图5D). ChIP分析显示,PRMT5招募到ST7标准启动子在转化的米诺中富集了3倍和5.5倍(P(P)=10−3)和JeKo(P(P)=0.006)MCL细胞系。与我们之前在NIH3T3细胞中的发现一致,BRG1招募到ST7标准启动子在Mino中增强了1.6倍和3倍(P(P)=10−2)和JeKo(P(P)=0.0007),而BRM招募不受影响。这些结果表明BRG1相关的PRMT5参与ST7标准转化B细胞中的转录抑制。由于PRMT5可以对称地甲基化H3R8和H4R3,我们评估了这些残基在ST7标准发起人(图5E). 我们发现H3R8的甲基化增加了1.7-(P(P)=0.013)和2.7倍(P(P)<10−4)分别在Mino和JeKo细胞中。类似地,H4R3的甲基化富集3.7-(P(P)=0.002)和2.5倍(P(P)<10−4)在这些MCL细胞系中。我们还分析了BRG1、BRM和PRMT5的招募以及H3R8和H4R3甲基化在ST7标准在另外两个淋巴瘤细胞系Daudi和Raji中发现了类似的结果(补充图3). 这些发现表明,PRMT5的表达增加直接参与ST7标准基因抑制、PRMT5募集以及H3R8和H4R3甲基化在转化的淋巴细胞系中发生改变。
PRMT5及其靶基因的表达ST7标准MCL临床样本中发生改变
我们发现PRMT5蛋白水平在多种转化的淋巴细胞中异常升高,包括Mino和JeKo MCL细胞系,因此ST7标准表达被抑制。为了证实我们的结果并评估从MCL患者分离的肿瘤中PRMT5和ST7之间是否存在反向关系,我们首先测量了PRMT5项目几种MCL临床样本的mRNA水平(补充图4). 我们的结果表明,MCL临床样本的PRMT5项目mRNA。此外,当我们分析MCL临床样本6和7中PRMT5蛋白的表达时,我们发现其在细胞核和胞质部分的水平升高(图6A). 更重要的是,免疫荧光分析显示,在MCL患者样本6和7中,PRMT5过度表达,H3R8和H4R3的整体对称甲基化成比例地增强(图6B). 因为PRMT5可以直接抑制ST7标准转录,我们分析ST7标准更多MCL患者样本中的mRNA和蛋白表达。实时RT-PCR显示ST7标准mRNA水平降低了2到30倍(P(P)<10−4)Western blot分析表明,ST7蛋白的表达在所有检测的MCL临床样本中均受到严重抑制(图7A和B). 此外,患者6和7的MCL样本中ST7蛋白的免疫荧光染色证实了Western blot结果,并表明ST7蛋白在MCL临床样本中的表达受到抑制(图7C). 接下来,我们分析了BRG1、BRM和PRMT5的招募ST7标准MCL患者样本6和7中的启动子(图7D). BRG1招募人数增加了2-(P(P)=0.002)至3.6倍(P(P)=0.006),而PRMT5招募增加了8-(P(P)=0.0002)至14倍(P(P)=0.003)。由于增加了BRG1相关PRMT5的招募ST7标准启动子,H3R8的对称甲基化被增强3-(P(P)=0.003)至7倍(P(P)=0.016),H4R3甲基化升高4.6-(P(P)=0.001)至5倍(P(P)=0.008)在MCL临床样本6和7中(图7E). 总之,这些结果表明,PRMT5表达的改变直接影响H3R8和H4R3的全球甲基化,并触发靶基因表达的变化,而靶基因表达又可能导致MCL。
图6。
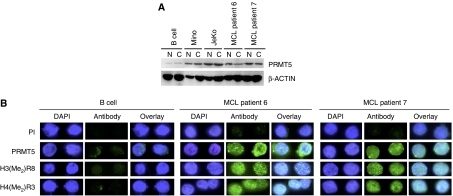
MCL患者PRMT5蛋白过度表达。(A类)正常CD19的核和细胞溶质提取物+使用抗PRMT5或对照β-肌动蛋白抗体通过Western blotting分析B细胞或MCL临床样本6和7(20μg)。(B类)用DAPI、PI或PRMT5、H3(Me2)R8和H4(Me2)第3段。
图7。
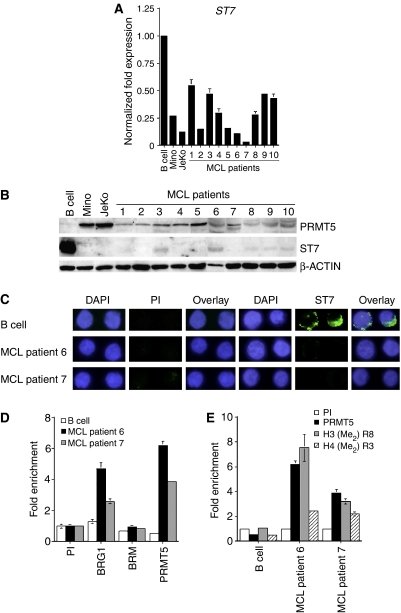
PRMT5的过度表达与ST7标准MCL临床样本中的沉默。(A类)实时RT-PCR分析ST7标准MCL患者样本1-10的mRNA表达。的表达式ST7标准使用规范化GAPDH公司作为内部控制。(B类)使用所示抗体对来自正常B细胞、Mino、JeKo和MCL临床样品1-10的20μg RIPA提取物进行Western blot分析。(C类)正常B细胞和MCL临床样本6和7细胞经DAPI、PI或免疫抗ST7抗体染色后的免疫荧光。照片是放大100倍拍摄的。(D类,E类)使用PI或指示的免疫抗体对来自正常B细胞和MCL临床样本6和7的交联染色质进行ChIP。通过实时PCR扩增免疫沉淀DNA,并计算每个抗体相对于PI样品的富集倍数。
敲除PRMT5改变转化淋巴细胞系的生长特性
先前的研究表明,PRMT5在NIH3T3细胞中的过度表达会导致过度增殖,而PRMT5的敲除会降低细胞的生长和增殖(朋友等, 2004). 由于在MCL细胞系和临床样本中PRMT5水平较高,我们研究降低PRMT5的水平是否会改变MCL细胞的生长。为了降低PRMT5的表达,用重组对照或反义(AS)-PRMT5慢病毒感染JeKo细胞,并在感染后的不同时间点评估PRMT5蛋白的表达(图8A). PRMT5蛋白水平在第4天显著降低,而β-肌动蛋白表达没有改变。当我们监测对照组和表达AS-PRMT5的JeKo细胞的增殖时,到第6天,生长明显下降了1.8倍(P(P)<10−4) (图8B). 为了评估细胞生长和增殖的减少是否是细胞死亡和/或细胞周期进展缓慢的直接结果,我们测量了对照细胞和AS-PRMT5感染细胞的DNA含量和BrdU掺入。我们的结果表明,AS-PRMT5细胞合并BrdU的效率比感染对照载体的亲代细胞低25%,并且其细胞周期曲线没有任何变化(数据未显示)。这些结果表明,减少PRMT5的表达会干扰转化的B细胞的增殖。
图8。
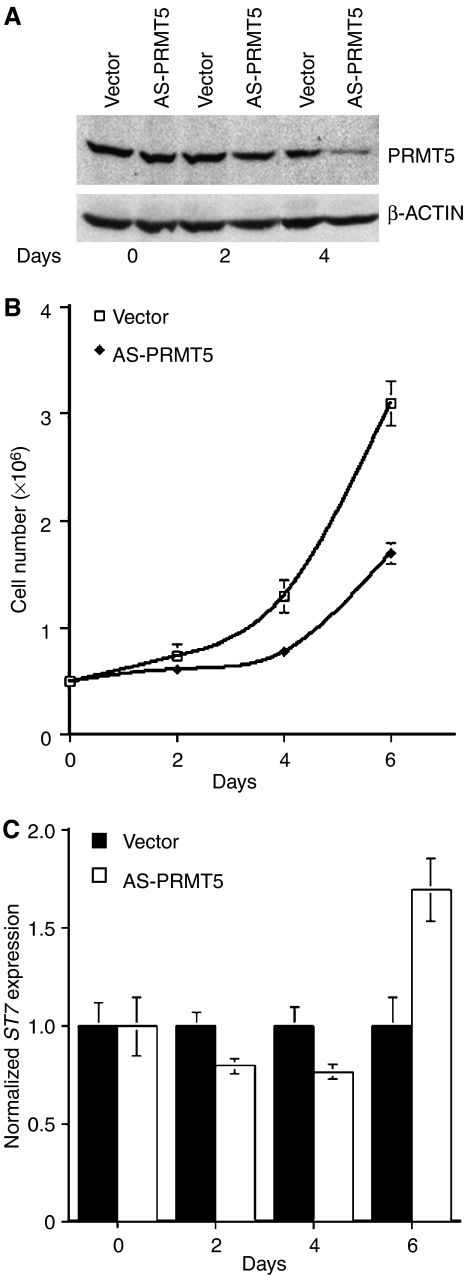
敲低PRMT5表达会影响转化的B细胞的生长。(A类)在用载体或AS-PRMT5慢病毒感染0、2和4天后,使用抗PRMT5和对照抗β-ACTIN抗体对20μg JeKo细胞RIPA提取物进行Western blot分析。(B类)感染控制载体或AS-PRMT5慢病毒的JeKo细胞增殖。每两天计数一次细胞,共6天,重复实验四次。(C类)ST7标准在指定时间通过实时RT-PCR评估慢病毒感染的JeKo细胞中的mRNA表达。ST7标准mRNA表达是相对于对照载体感染的JeKo细胞来表示的,并且标准化为GAPDH公司.
我们已经证明,PRMT5可以直接调节ST7标准转录。因此,我们测量ST7标准通过实时RT-PCR检测对照组和表达AS-PRMT5的JeKo细胞的mRNA水平(图8C). 我们的结果表明,随着PRMT5蛋白水平在第6天下降ST7标准诱导1.7倍(P(P)=10−3). 尽管有这种转录去表达,ST7蛋白水平没有增加,这表明可能有其他机制参与调节ST7蛋白表达(数据未显示)。对细胞增殖和ST7标准当PRMT5在Raji细胞中被敲除时,观察到mRNA表达(补充图5). 这些发现提供了更多证据,证明敲低癌B细胞中PRMT5的表达会影响其生长特性,并且ST7标准表达是转录后调控的。
讨论
染色质的表观遗传修饰是密集研究的重点,因为需要解决癌症中许多尚未解决的问题。了解组蛋白翻译后的变化如何影响基因表达,以及该过程的扰动如何导致疾病,必将为癌症治疗提供新的途径。我们以前的研究表明,PRMT5在永生化而非转化的成纤维细胞中的过度表达可诱导其过度增殖和锚定非依赖性生长,这强烈表明PRMT5对肿瘤的发生起着重要作用(朋友等, 2004). 在本研究中,我们发现PRMT5蛋白在多种人类淋巴癌细胞系中诱导表达,包括患者衍生的MCL细胞系以及MCL临床样本。我们还将PRMT5的错误表达与miR-92b和miR-96的异常表达联系起来,并表明这些变化的结果是H3R8和H4R3的整体对称甲基化增加。更具体地说,我们证明了PRMT5靶基因的表达,ST7标准在PRMT5、miR-92b和miR-96表达改变的人类淋巴癌细胞系中被抑制。此外,我们的研究还表明,敲低PRMT5表达会减少转化淋巴细胞系的增殖,这表明PRMT5水平的降低对正常细胞生长至关重要。
miR-92b和miR-96的异常表达与MCL中PRMT5翻译增强有关
对B细胞、转化淋巴细胞系和MCL临床样本的核和细胞溶质提取物的分析表明,PRMT5蛋白在两个腔室中的表达均显著升高(图1和6). 我们还分析了PRMT5蛋白在各种实体瘤细胞系中的表达,包括胶质瘤(U251,Gli3605)、腺癌(HeLa S3,SW13)、乳腺癌和肺癌(BT549,A549)以及肝癌(HepG2)(Pal和Sif,未发表),我们发现,与NIH3T3、Rat1a和PC12细胞系相比,PRMT5蛋白在细胞核和胞质溶胶中的整体表达增加。核PRMT5与染色质重塑相关,包括基于BRG1/BRM的hSWI/SNF和NURD复合物,并参与细胞周期调节器和肿瘤抑制基因的转录抑制(朋友等, 2003,2004;盖森内克等, 2006). 因此,当PRMT5水平因过度表达或转化癌细胞中的情况而升高时,可能会抑制关键靶基因的转录并促进肿瘤发生。
我们的研究结果表明,PRMT5在转化的Mino和JeKo MCL细胞系中的表达增加是增强的直接结果PRMT5项目mRNA翻译(图2和3A). 更令人惊讶的是,两个miRNAs的表达预测与PRMT5项目患者源性Mino和JeKo MCL细胞以及MCL临床样本中的3′UTR降低,两种不同淋巴瘤细胞系中miR-92b和miR-96的重新表达降低了PRMT5的翻译体内(图4A). 此外,miR-92b或miR-96种子序列的突变消除了PRMT5翻译抑制体内表明观察到的miR-92b和miR-96的抑制作用是特异性的。我们试图重述PRMT5的翻译抑制在体外使用野生型miR-96时未成功(未显示数据)。我们已经弥补了PRMT5项目mRNA:miR杂交分子通过增加种子序列以外的互补性,我们已经能够特异性抑制PRMT5翻译在体外(图4B). 进一步支持miR-92b和miR-96参与调节PRMT5翻译,这是因为我们观察到PRMT5 3′UTR可以增强转化的JeKo细胞中的荧光素酶翻译,而miR-92b+miR-96表达较低(图4C). 野生型miR-92b或miR-96的转染导致JeKo和Raji细胞中PRMT5表达的有效降低(图4A)发现miR-92b或miR-96结合位点的突变提高了荧光素酶的翻译(图4C),认为miR-92b和miR-96在调节PRMT5蛋白表达中起着重要作用。
PRMT5表达增加增强了H3R8和H4R3的整体对称甲基化,并伴有ST7的抑制
根据我们以前的工作和目前的观察,PRMT5与染色质重塑物的结合增强了其组蛋白甲基转移酶活性,并赋予其影响靶基因表达的能力,从而最终促进细胞生长和转化(朋友等, 2004). 由于PRMT5优先靶向组蛋白H3R8和H4R3,我们分析了它们在转化MCL细胞中的甲基化状态,并表明这些位点在患者来源的MCL细胞系和MCL临床样本中高度甲基化(图1C和6B). 更具体地说,当我们分析组蛋白甲基化ST7标准启动子,我们发现H3R8和H4R3都是高甲基化的(图5和7). 鉴于很明显,PRMT5的招募ST7标准启动子增强H3R8和H4R3甲基化,组蛋白H3R8/H4R4甲基化水平与ST7标准转录抑制(图5和7).
众所周知,组蛋白的不同翻译后修饰可以协同作用或拮抗作用来指定转录结果(菲施勒等, 2003). 例如,组蛋白H3K9和H3K27三甲基化标记在受抑制的超双胸发起人(林格罗斯等, 2004). 因此,在ST7标准转录,还有其他表观遗传标记参与其调控,这些标记可能因细胞系和临床样本而异。第二种可能性是H3R8/H4R3甲基化与ST7标准转录抑制可能是由于对称甲基化H3R8和H4R3结合蛋白表达的差异引起的。最近的研究清楚地表明,甲基化组蛋白可以被特定的甲基结合蛋白识别和结合,这对转录调控很重要。WDR5蛋白就是一个很好的例子,它可以与二甲基化的H3K4结合并支持转录激活HOXC8型和HP1,HP1可以识别三甲基化的H3K9并诱导沉默(霍尔等2002年;威索卡等, 2005). 因此,重要的是要确定是否存在额外的沉默表观遗传标记ST7标准启动子,以及是否有特定因子可以对称地结合甲基化的H3R8和H4R3。
我们已经确定ST7标准在转录和翻译水平上都受到调控。敲低转化JeKo和Raji细胞中PRMT5的表达诱导ST7标准转录,但不影响ST7蛋白表达(图8C和补充图2C和5C). 同样,转化的淋巴细胞表达低水平的ST7标准mRNA未显示任何可检测到的ST7蛋白水平(图5和7). 我们已经确定miR-92b和miR-96的表达减少会导致PRMT5翻译增强。因此,我们不能排除ST7标准-特异性miRNA可能抑制ST7的翻译。需要更多的实验来检测ST7-特异性miRNAs在正常和转化的B细胞中的表达。
PRMT5在套细胞淋巴瘤中的作用
越来越明显的是,组蛋白修饰酶的错误表达和/或突变与癌症病因学有关。例如,混合系白血病基因MLL1甲基化H3K4并激活转录,在急性白血病中经常移位,因此产生50多种不同的白血病MLL融合蛋白(Slany,2005年). 大多数MLL融合蛋白通过增加HOX公司通过组蛋白高乙酰化和H3K79甲基化机制实现基因表达(斯莱尼,2005年). 另一种似乎在各种癌症中发生改变的表观遗传标记涉及H3K27特异性甲基转移酶EZH2,它与基因沉默密切相关。EZH2在前列腺癌、乳腺癌和胃癌中的过度表达已被证实,并且似乎与肿瘤的高度侵袭性相关(瓦兰巴利等2002年;克莱尔等, 2003;松川等, 2006). 此外,EZH2在Ramos淋巴瘤和前列腺癌细胞系中的表达增加可诱导其生长和过度增殖(维瑟牌汽车等, 2001;瓦兰巴利等2002年). 组蛋白精氨酸甲基化在肿瘤发生中的作用尚不清楚。
支持PRMT5在癌症中作用的证据来自研究,研究表明PRMT5经常在胃癌中上调(基姆等, 2005). 此外,当用ER-α转染MDA-MB-231乳腺癌细胞系并用17β-雌二醇处理时,细胞增殖减少,伴随着PRMT5表达减少(莫格斯(Moggs)等, 2005). 我们的结果表明,PRMT5在淋巴源性癌细胞(包括MCL临床样本)中发生翻译上调。结果,H3R8和H4R3的对称甲基化增强。组蛋白精氨酸甲基化增强的影响是基因表达调控不当,在PRMT5靶基因的情况下,ST7标准,存在明显的转录抑制,这似乎与癌细胞生长有关。我们还发现,敲低MCL和Burkitt淋巴瘤细胞系中PRMT5的表达会降低细胞增殖。因此,PRMT5似乎是一种关键的组蛋白修饰酶,通过组蛋白精氨酸甲基化调节靶基因的表达来控制细胞生长。
PRMT5的高表达可以影响组蛋白H3R8和H4R3甲基化以外的其他途径。最近,发现PRMT5与与NURD复合物相关的MBD2相互作用并甲基化(盖森内克等, 2006). 甲基化的结果是MBD2与甲基化DNA的结合减少,无法抑制转录(Tan和Nakielny,2006年). 因此,除了组蛋白修饰外,PRMT5可能通过负向调节基于MBD2的NURD复合体的活性来促进肿瘤发生。因此,研究过度表达PRMT5的癌细胞中MBD2的甲基化状态,并验证这些癌细胞中NURD靶基因的表达是否发生改变,将是一件有趣的事情。此外,我们的研究结果表明,减少PRMT5的表达可以抑制细胞增殖,这为设计旨在解决靶向PRMT5治疗相关性的新策略提供了一个良好的起点。
材料和方法
细胞培养、B细胞分离、转染、荧光素酶分析、慢病毒产生和细胞感染
正常和转化的淋巴细胞在补充有10-20%FBS的RPMI-1640中培养。正常B细胞从扁桃体中分离,通过合作人体组织网络(CHTN)从儿童医院获得,如补充数据根据IRB批准且符合HIPPA的方案收集正常和转化的人类B淋巴细胞。有关转染、荧光素酶分析、慢病毒产生和细胞感染的详细信息,请参阅补充数据.
抗体、蛋白质印迹和免疫荧光分析
有关详细信息,请参阅补充数据.
逆转录,实时聚合酶链式反应,核运行分析,多核糖体分析,核糖核酸酶保护分析(RPA),在体外转录、加盖、聚腺苷酸化和翻译
详细协议如所述补充数据.
染色体免疫沉淀分析法
ChIP实验使用约1×10的可溶性交联染色质进行7正常或转化的B细胞如前所述,但用混合胶束缓冲液清洗两次(朋友等, 2003,2004). 评估招聘ST7标准启动子,在10μl反应中对3μl洗脱DNA进行实时PCR,如补充数据.
统计分析
为了统计验证不同组内多个样本产生的数据,使用方差分析(ANOVA)计算P(P)-值。为了确定两组之间差异表达的基因或不同染色质重塑的招募吨-测试用于计算P(P)-值。在所有情况下,GraphPad Prism4软件都用于生成P(P)-值。
致谢
我们感谢L Comai提供慢病毒表达载体,D Schoenberg和E Murray帮助制备多核糖体,并提供质粒pCMV-LUC,T Ryan在扁桃体B细胞分离方面提供技术帮助,P Porcu和D Lucas提供MCL临床样本。我们还感谢D Dakhlallah、S Goel和L Wang对手稿的批判性阅读。这项工作得到了美国国家癌症研究所拨款RO1 CA116093和PO1 CA101956,以及美国癌症协会向SS拨款RSG-0418201-GMC的支持。
工具书类
-
Ancelin K、Lange UC、Hajkova P、Schneider R、Bannister AJ、Kouzaride T、Surani MA(2006)Blimp1与Prmt5相关,并指导小鼠生殖细胞中的组蛋白精氨酸甲基化。自然细胞生物学8:623–630[内政部] [公共医学] [谷歌学者]
-
Bedford MT,Richard S(2005)精氨酸甲基化是蛋白质功能的新兴调节器。分子细胞18:263–272[内政部] [公共医学] [谷歌学者]
-
Cao R,Wang L,Wang H,Xia L,Erdjument-Bromage H,Tempst P,Jones RS,Zhang Y(2002)组蛋白H3赖氨酸27甲基化在多梳群沉默中的作用。科学298:1039–1043[内政部] [公共医学] [谷歌学者]
-
Dacwag CS、Ohkawa Y、Pal S、Sif S、Imbalzano AN(2007)精氨酸甲基转移酶Prmt5蛋白是肌发生所必需的,因为它促进ATP依赖的染色质重塑。分子细胞生物学27:384–394[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Esteller M(2006)表观遗传学提供了新一代致癌基因和肿瘤抑制基因。英国癌症杂志94:179–183[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Fabbrizio E、El Messaoudi S、Polanowska J、Paul C、Cook JR、Lee JH、Negre V、Rousset M、Pestka S、Le Cam A、Sardet C(2002)II型精氨酸甲基转移酶PRMT5对转录的负调控。EMBO代表3:641-645[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Fischle W,Wang Y,Allis CD(2003)组蛋白和染色质串扰。Curr Opin细胞生物学15:172–183[内政部] [公共医学] [谷歌学者]
-
Friesen WJ、Paushkin S、Wyce A、Massenet S、Pesiridis GS、Van Duyne G、Rappsilber J、Mann M、Dreyfuss G(2001)甲基体,一种含有JBP1和pICln的20S复合物,产生二甲基精氨酸修饰的Sm蛋白。摩尔细胞生物学21:8289–8300[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Guezennec XL、Vermeulen M、Brinkman AB、Hoeijmakers WA、Cohen A、Lasonder E、Stunneberg HG(2006)MBD2/NuRD和MBD3/NuRD是两种不同的复合物,具有不同的生化和功能特性。分子细胞生物学26:843–851[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Hake SB,Xiao A,Allis CD(2004)将共价组蛋白修饰的表观遗传“语言”与癌症联系起来。英国癌症杂志90:761–769[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Hall IM,Shankararayana GD,Noma K,Ayoub N,Cohen A,Grewal SI(2002)异染色质结构域的建立和维护。科学297:2232–2237[内政部] [公共医学] [谷歌学者]
-
Hosohata K,Li P,Hosohaat Y,Qin J,Roeder RG,Wang Z(2003)一种参与雄激素受体依赖性转录的新型复合物的纯化和鉴定。分子细胞生物学23:7019–7029[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Kim JM,Sohn HY,Yoon SY,Oh JH,Yang JO,Kim JH,Song KS,Rho SM,Yoo HS,Kim YS,Kin JG,Kim NS(2005)使用包含胃癌细胞中表达的新表达序列标签的cDNA微阵列鉴定胃癌相关基因。临床癌症研究11:473–482[公共医学] [谷歌学者]
-
Kleer CG、Cao Q、Varambally S、Shen R、Ota I、Tomlins SA、Ghosh D、Sewalt RG、Otte AP、Hayes DF、Sabel MS、Livant D、Weiss SJ、Rubin MA、Chinnaiyan AM(2003)EZH2是侵袭性乳腺癌的标志物,促进乳腺上皮细胞的肿瘤转化。美国国家科学院院刊100:11606–11611[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Kwak YT,Guo J,Prajapati S,Park KJ,Surabhi RM,Miller B,Gehrig P,Gaynor RB(2003)SPT5的甲基化调节其与RNA聚合酶II的相互作用和转录延伸特性。摩尔细胞11:1055–1066[内政部] [公共医学] [谷歌学者]
-
Martin C,Zhang Y(2005)组蛋白赖氨酸甲基化的多种功能。Nat Rev Mol细胞生物学6:838–849[内政部] [公共医学] [谷歌学者]
-
Matsukawa Y、Semba S、Kato H、Ito A、Yanagihara K、Yokozaki H(2006)zeste同源物2增强子的表达与人类胃癌预后不良相关。癌症科学97:484–491[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Milne TA、Briggs SD、Brock HW、Martin ME、Gibbs D、Allis CD、Hess JL(2002)MLL将SET域甲基转移酶活性靶向霍克斯基因启动子。分子电池10:1107–1117[内政部] [公共医学] [谷歌学者]
-
Moggs JG,Murphy TC,Lim FL,Moore DJ,Stuckey R,Antrobus K,Kimber I,Orphanides G(2005)雌激素在重新表达ERalpha的乳腺癌细胞中的抗增殖作用是由细胞周期基因的异常调节介导的。分子内分泌杂志34:535–551[内政部] [公共医学] [谷歌学者]
-
Pal S、Vishwanath SN、Erdjument-Bromage H、Tempst P、Sif S(2004)《人类SWI/SNF-associated PRMT5甲基化组蛋白H3精氨酸8》,负调控ST7和NM23抑癌基因的表达。分子细胞生物学24:9630–9645[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Pal S,Yun R,Datta A,Lacomis L,Erdjument-Bromage H,Kumar J,Tempst P,Sif S(2003)mSin3A/组蛋白脱乙酰酶2-和含有PRMT5-的Brg1复合物参与Myc靶基因cad的转录抑制。分子细胞生物学23:7475–7487[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Papp B,Muller J(2006)组蛋白三甲基化和通过trxG和PcG蛋白维持转录ON和OFF状态。基因Dev 20:2041–2054[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Pollack BP、Kotenko SV、He W、Izotova LS、Barnoski BL、Pestka S(1999)酵母蛋白Skb1和Hsl7p的人类同源物与Jak激酶相互作用,并含有蛋白甲基转移酶活性。生物化学杂志274:31531–31542[内政部] [公共医学] [谷歌学者]
-
Ringrose L,Ehre H,Paro R(2004)组蛋白H3赖氨酸9和27甲基化对多梳复合物局部特异性稳定性的显著贡献。分子细胞16:641–653[内政部] [公共医学] [谷歌学者]
-
Santos-Rosa H,Caldas C(2005),染色质修饰酶,组蛋白代码与癌症。《欧洲癌症杂志》41:2381–2402[内政部] [公共医学] [谷歌学者]
-
Sif S(2004)ATP依赖的核小体重塑复合物:专为处理染色质而设计的酶。细胞生物化学杂志91:1087–1098[内政部] [公共医学] [谷歌学者]
-
Slany RK(2005)当表观遗传学杀死:白血病中的MLL融合蛋白。血醇癌23:1–9[内政部] [公共医学] [谷歌学者]
-
Tan CP,Nakielny S(2006)通过蛋白质精氨酸甲基化控制DNA甲基化系统成分MBD2。分子细胞生物学26:7224–7235[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
Varambally S、Dhanasekaran SM、Zhou M、Barrette TR、Kumar-Shinha C、Sanda MG、Ghosh D、Pienta KJ、Sewalt RG、Otte AP、Rubin MA、Chinnaiyan AM(2002)多梳组蛋白EZH2参与前列腺癌的进展。自然419:624–629[内政部] [公共医学] [谷歌学者]
-
Visser HP、Gunster MJ、Kluin-Nelemans HC、Manders EM、Raaphorst FM、Meijer CJ、Willemze R、Otte AP(2001)Polycomb组蛋白EZH2在增殖培养的人类外套细胞淋巴瘤中上调。英国血液学杂志112:950–958[内政部] [公共医学] [谷歌学者]
-
Wang B,Love TM,Call ME,Doench JG,Novina CD(2006)《短RNA定向翻译基因沉默综述》在体外.分子细胞22:553–560[内政部] [公共医学] [谷歌学者]
-
Witzig TE(2005)外套细胞淋巴瘤的当前治疗方法。临床肿瘤学杂志23:6409–6414[内政部] [公共医学] [谷歌学者]
-
Wysocka J、Swigut T、Milne TA、Dou Y、Zhang X、Burlingame AL、Roeder RG、Brivanlou AH、Allis CD(2005)WDR5与在K4甲基化的组蛋白H3相关,对H3 K4甲基化和脊椎动物发育至关重要。手机121:859–872[内政部] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。

