p53抑癌基因调节特定靶基因的转录。正常情况下,p53的水平受到MDM2蛋白的严格控制,MDM2蛋白质受p53上调,靶向p53进行蛋白酶体介导的降解。p53在细胞应激反应中积累,例如DNA损伤、癌基因激活和缺氧。活化的p53诱导或抑制参与细胞周期停滞、DNA修复、凋亡和衰老的基因的转录,所有这些对于维持基因组稳定性和/或消除早期肿瘤细胞都很重要。在p53诱导的基因中,p21是细胞周期阻滞的效应器,而Bax、PUMA等是p53依赖性凋亡的介导者(1). p53介导的Bcl-2、IGF1受体和端粒酶(hTERT)的反式阻遏对凋亡反应也很重要(2——4). 转录转录激活是通过p53与靶基因中的特定基序结合而发生的,而转录抑制可能涉及与其他蛋白质的相互作用。p53还可以通过转录依赖机制促进细胞死亡(5). 事实上,p53可以转移到线粒体并与抗凋亡蛋白相互作用以促进凋亡(6).
将近一半的人类肿瘤携带突变型p53(参见网址:http://p53.free.fr和网址:www-p53.iarc.fr). p53失活导致p53靶基因调控不当,无法触发p53依赖性反应。p53缺失小鼠在早期容易发生自发性淋巴瘤和肉瘤(7). p53的转录激活对p53诱导的凋亡至关重要,而凋亡是肿瘤发展的重要障碍(8,9).
为了更好地理解p53通路和p53依赖性肿瘤抑制,有必要进一步了解已知p53靶基因的功能,并确定新的p53靶点。一些研究通过微阵列分析解决了p53依赖性基因表达(10,11). 这些研究表明,p53可以诱导广泛的靶基因,这些靶基因除了参与细胞周期阻滞和凋亡外,还参与多种细胞过程,许多基因受到p53的抑制(12). 最近的一项研究使用基于ChIP的方法鉴定了新的p53靶基因(13). 与这些研究一致,超过4800个基因被发现包含至少一个p53基序(14). 因此,p53可以潜在地调节许多基因,尽管不一定在同一个细胞中对特定类型的压力作出反应。
到目前为止,已经在mRNA水平上进行了p53依赖性基因表达模式的研究。然而,由于蛋白质编码基因的功能是由蛋白质产物执行的,因此在蛋白质水平上研究p53依赖性表达很重要。这可能揭示新的p53靶点,包括受转录依赖机制调节的靶点,例如通过翻译后修饰和/或改变蛋白质稳定性。在这里,我们使用携带野生型p53和等基因p53缺失细胞的HCT116结肠癌细胞分析p53调节蛋白组(15)、窄范围2D凝胶电泳(2DE)和质谱。我们确定了参与mRNA处理、翻译、转移和凋亡等过程的p53靶点。
结果
丝裂霉素C(MMC)激活p53和下游靶点。
为了激活内源性p53,我们用10μg/ml MMC处理HCT116结肠癌细胞,MMC是一种交联DNA的DNA损伤剂,阻止了两条DNA链的分离。免疫染色显示,与MMC孵育8小时后,wtp53中的p53诱导+/+单元格(图1 A类). 为了证实p53靶基因的激活,我们分析了MMC处理的p53+/+和p53−/−Western blotting检测细胞。在p53中观察到Ser-15诱导p53和p53磷酸化+/+但不在p53中−/−细胞。我们还检测到p21和MDM2的上调,这两个经典的p53靶点和p53靶Wig-1(图1B类). 此外,p53重表达的人类端粒酶逆转录酶(hTERT)基因在p53中的表达降低+/+单元格(图1B类). FACS分析证实p53中细胞死亡比例较高+/+单元格(图1C).
图1。
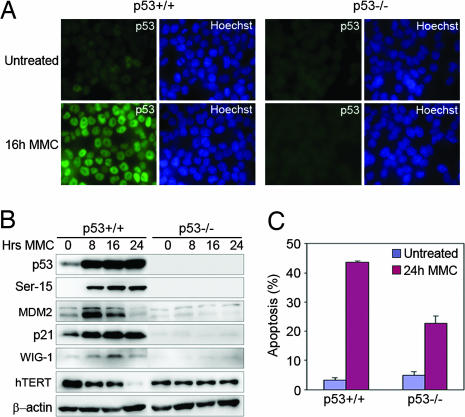
MMC处理HCT116细胞中p53激活和诱导凋亡。(A类)HCT116的p53免疫染色+/+和p53−/−用10μg/ml MMC处理24小时后的细胞(B类)MMC处理HCT116 p53蛋白的Western blot分析+/+和p53−/−细胞表现出对p53的诱导和对几个p53靶点的调节。(C)通过碘化丙啶(PI)染色和FACS分析评估细胞凋亡。
质谱法鉴定55种差异表达蛋白质。
MMC处理的p53总蛋白+/+或p53−/−对时间点0、8、16和24小时的HCT116细胞进行2DE。在四个匹配集(pH 3.9-5.1;4.7-5.9;5.5-6.7;和6.3-8.3)中,PDQuest软件(Bio-Rad,Hercules,CA)分析(数据未显示)分离并检测到多达5789个蛋白质点。匹配集II(pH4.7–5.9)和匹配集III(pH5.5–6.7)包含所有分离点的2/3。我们发现115个蛋白点在表达水平上表现出统计上的显著差异(P(P)< 0.05; Mann–Whitney试验)。统计学上显著的变化被定义为与p53阴性对照细胞中的蛋白表达水平相比,表达水平在至少两个时间点上至少增加或减少1.5倍。为了评估再现性,计算了三个重复凝胶之间的相关系数。平均值第页该值为0.89(范围:0.86–0.93),表明高质量的2DE凝胶以及稳定和可重复的培养和处理条件。我们通过质谱法鉴定了55种p53调节多肽[支持信息(SI)表2]. 其中42个蛋白以p53依赖性方式上调,13个蛋白下调。MMC诱导的蛋白在p53中增加了100倍或更多+/+而在p53阴性细胞中,它们的表达水平基本不变,甚至下降。在下调蛋白中,p53的表达降低了10倍+/+细胞,没有诱导或在某些情况下p53略有增加−/−单元格(SI表2). 一些已鉴定的蛋白质列于表1.
表1。
| 蛋白质 |
表情,折叠变化
|
推定结合位点 |
第53页+/+
|
第53页−/−
|
| 8小时 |
16小时 |
24小时 |
8小时 |
16小时 |
24小时 |
| 上调监管 |
| 半胱氨酸天冬氨酸蛋白酶3 |
4.19 |
6.41 |
— |
— |
— |
— |
是的 |
| eIF5A型 |
1.94 |
11.26 |
13.01 |
— |
— |
1.42 |
是的 |
| hnRNP C1/C2 |
2.18 |
2.58 |
3.49 |
— |
— |
— |
不 |
| hnRNP K公司 |
7.67 |
8.3 |
— |
— |
— |
— |
是的 |
| 拉明A/C |
3.69 |
67.2 |
133.7 |
3.7 |
11.1 |
18 |
是的 |
| 编号23-H1 |
1.46 |
2.21 |
1.38 |
— |
— |
— |
是的 |
| 核蛋白 |
1.49 |
1.66 |
— |
— |
— |
— |
是的 |
| TAT结合蛋白1 |
— |
1.48 |
13.43 |
— |
— |
— |
是的 |
| 硫氧还蛋白 |
1.43 |
8.11 |
11.03 |
1.3 |
1.7 |
1.77 |
不 |
| 14-3-3 σ |
— |
2.06 |
6.07 |
— |
— |
— |
是的 |
| 下调监管 |
| hnRNP K,亚型a |
−1.54 |
−3.69 |
−10.28 |
— |
— |
— |
是的 |
| hnRNP C1/C2一个 |
— |
−1.67 |
−4.96 |
— |
1.32 |
1.39 |
不 |
| PP2A型 |
−1.5 |
−2.3 |
— |
— |
— |
— |
是的 |
| Prx II公司 |
— |
−1.56 |
−2.85 |
— |
— |
— |
不 |
| TrpRS公司 |
−1.3 |
−1.9 |
−2.71 |
— |
— |
— |
是的 |
七种已鉴定蛋白质的表达变化。
5个已鉴定的上调点、真核翻译因子5A(eIF5A)、异质核核糖核蛋白C1/C2(hnRNP C1/C2)、异质核糖核蛋白质K(hnRNP-K)、层粘连蛋白A/C和转移抑制因子Nm23-H1的表达变化以及2个已鉴定出的下调点过氧化物酶原II(Prx II)和色氨酸-tRNA合成酶(TrpRS),如图所示图2如eIF5A所示,PDQuest软件的3D渲染功能用于可视化蛋白质斑点,并将两个或多个重叠斑点作为单个蛋白质的误解降至最低(图2B类). 计算所有匹配集和三个单独实验中所有点的光密度,并将其编译成直方图,以显示不同时间点的蛋白质表达(图3 A–I类). 我们检测到,与0小时的水平相比,24小时eIF5A蛋白水平的p53依赖性增加了10倍以上。hnRNP K水平在8小时时已经显著诱导,在16小时时保持较高水平,并在24小时下降到与未经处理的细胞相当的水平。Lamin A/C在治疗前从几乎无法检测到的水平诱导至16小时的稳定表达水平,24小时甚至更高水平。Nm23-H1水平在16小时达到峰值,而hnRNP C1/C2在24小时达到最高水平。24小时后,Prx II和TrpRS蛋白的水平逐渐降低至未处理细胞水平的25-30%。在所有情况下,我们都没有观察到p53缺失细胞的主要表达变化(图3 A–G). 此外,我们确定了两个下调点,即hnRNP K亚型a和hnRNP C1C/2 a(图3 H(H)和我). 这些斑点可能代表这些蛋白质的特定亚型和/或由于转录后修饰而产生的不同流动性(参见讨论).
图2。
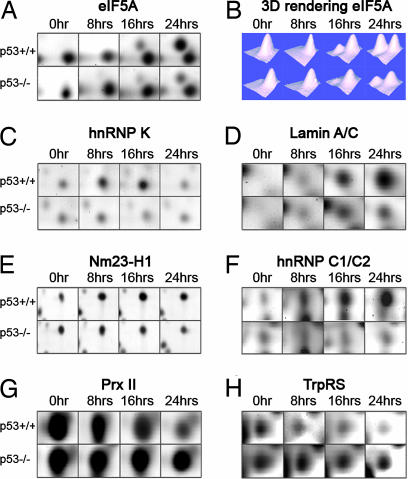
7种已鉴定蛋白的p53依赖性表达。(A类,C–H型)凝胶切片显示四个时间点的指示蛋白点的差异表达。(B类)PDQuest软件对eIF5A斑点进行3D渲染分析。
图3。
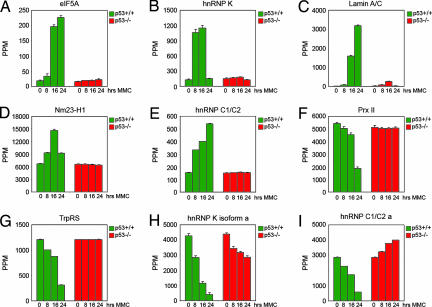
p53调节蛋白表达。通过PDQuest软件分析量化指示斑点的光密度。图表显示了三次独立凝胶运行的平均值。
验证p53在蛋白质和RNA水平上的依赖性调节。
为了验证这些结果,我们使用针对七种已鉴定蛋白质的抗体进行了Western blot分析。结果与2DE数据基本一致(图4). 然而,在蛋白质印迹分析中,hnRNP K和层粘连蛋白A/C的诱导不太明显,这可能是因为2D凝胶上出现了特异性调节的同种型。此外,与0小时表达的水平相比,我们没有检测到Prx II的显著下调。Western blot分析显示,PrxⅡ在8小时表达短暂增加,在16和24小时表达低水平+/+而在MMC处理后的所有时间点,p53阴性细胞中的Prx II水平均升高。有趣的是,MMC处理的p53中检测到一种快速迁移的eIF5A蛋白物种+/+单元格(图4)这表明一种特定的亚型受p53调控。Western blotting显示两种TrpRS蛋白;而53 kDa基因在p53中表达下调+/+细胞中,70kDa的物种在表达上没有表现出任何变化(图4).
图4。
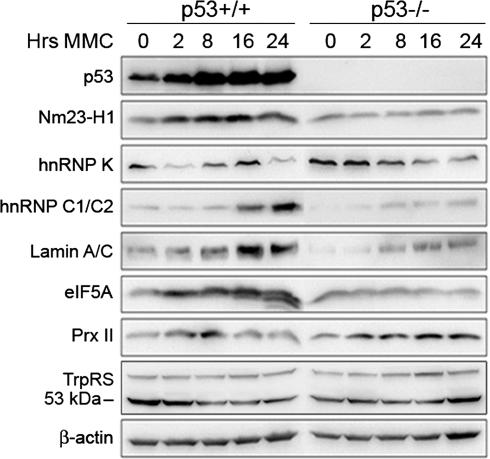
MMC治疗HCT116 p53在指定时间点的p53依赖性蛋白表达的Western blot分析+/+和p53−/−细胞。β-肌动蛋白作为负载对照。
我们还通过定量实时RT-PCR检测了每种蛋白质的mRNA水平(图5 A–G). 已知的p53靶点p21、MDM2和Wig-1被纳入阳性对照(图5 H(H)和数据未显示)。我们观察到MMC治疗的p53中eIF5A、hnRNP K、层粘连蛋白A/C和Nm23-H1以及所有三个对照的mRNA水平增加+/+单元格(图5 A–D与p53的转录调控一致。在不同时间点,mRNA水平与蛋白质水平表现出良好的相关性。相反,无论p53状态如何,MMC处理细胞中hnRNP C1/C2的mRNA水平均显著降低,尤其是在16和24小时时,这表明观察到的hnRNP C1/C2蛋白表达水平的p53依赖性增加不是由于转录调节所致(图5E类). 同样,我们对Prx II和TrpRS mRNA水平的分析显示,mRNA和蛋白质水平的调节不一致。Prx II mRNA水平没有变化,除了在MMC治疗16小时时p53中有轻微增加+/+细胞,而p53蛋白在24小时时显著减少+/+细胞,根据2DE分析。在MMC处理的细胞中,TrpRS mRNA仅略有下降,表明观察到的p53依赖性下调发生在蛋白水平(图5 F类和G公司).
图5。
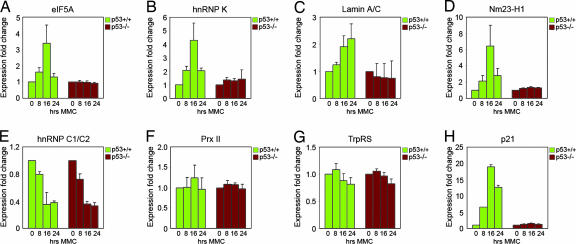
p53的转录调控。(A–G)MMC治疗HCT116 p53中指定时间点指定基因p53依赖性mRNA表达的实时PCR分析+/+和p53−/−细胞。(H(H))p53靶点p21作为阳性对照。
对另外两种野生型p53癌细胞系MCF7和U2OS进行的Western blot分析显示,MMC处理后,蛋白表达发生了类似的变化,证实所观察到的变化并不是HCT116细胞的独特特征(SI图6).
蛋白质表达变化与细胞凋亡的关系。
确定观察到的表达变化是否与凋亡而非p53激活有关就其本身而言,我们检测了MMC处理的HCT116细胞在caspase抑制剂zVAD存在下的蛋白质水平。即使在zVAD存在的情况下,hnRNP C1/C2、层粘连蛋白A/C和Nm23-H1也以p53依赖的方式诱导。同样,eIF5A也被诱导,尽管程度较低(SI图7).
我们还检测了用staurosporine(STS)处理的HCT116细胞中的蛋白表达,STS是一种触发p53诱导的依赖性凋亡的药物。eIF5A、hnRNP C1/C2、层粘连蛋白A/C和Nm23-H1均未被STS诱导。这些结果表明,这里测试的大多数鉴定蛋白并不是由凋亡诱导的,而是直接的p53靶点(SI图8).
已鉴定蛋白质编码基因的启动子分析。
我们使用p53MH算法分析了编码p53结合基序的已识别蛋白质的基因中的启动子区域,包括转录起始点上游的5kb以及外显子1和内含子1(16). 24个编码上调蛋白的基因和8个编码下调蛋白的基因包含至少一个假定的p53结合位点,与p53共有结合基序的最小相似性为80%(SI表2). 这些结果支持这样的观点,即许多已鉴定的p53诱导蛋白确实是真正的p53转录靶点。这些包括,例如,eIF5A、hnRNP K、Nm23-H1、膜联蛋白1和核磷蛋白(NPM)。然而,一些已鉴定的基因似乎缺乏p53结合基序,这表明它们不是经典的p53转录靶点。
讨论
p53可能通过转录依赖和转录依赖机制调节下游靶点。转录依赖性效应可以是直接或间接的,即由p53本身或p53的一个或多个转录靶点介导。也可以想象,p53通过影响特定蛋白质的稳定性、细胞内定位和/或活性,以转录依赖的方式直接调节特定蛋白质。这种调节可能涉及蛋白质-蛋白质相互作用和翻译后修饰,如磷酸化和泛素化。p53调节蛋白表达的分析应提供有关转录依赖性和转录依赖性调节的信息。在这里,我们使用高分辨率micropH2DE和质谱法对p53调节的蛋白质组进行了全面分析。分离出接近5800个蛋白点,其中115个点的表达水平发生了显著的p53依赖性变化。因此,本研究中约2%的分离蛋白点受到p53的影响。这一结果与先前的微阵列研究一致,该研究表明p53可能调节所有人类基因的2-4%(11,12)并且表明受p53调控的蛋白质数量并没有大大超过p53调控基因的数量。然而,在使用小鼠温度敏感型p53进行的DNA微阵列分析中,放线菌酮对蛋白质合成的抑制导致p53调节转录物的比例从5.5%降至0.88%(10). 这支持了这样一种观点,即许多已确定的p53调节基因是间接靶点。
我们确定了经典的p53靶点14-3-3σ和caspase-3(表1),证实了我们的实验方法是有效的。然而,其他已知的p53靶点,如p21、MDM2、Bax和PUMA,不在已鉴定的蛋白质中。这可能是因为2DE的分辨率和灵敏度不允许检测所有细胞蛋白。特定蛋白质的检测将取决于丰度、分子量、等电点和其他因素。
这里确定的p53调节蛋白属于不同的功能类别,包括mRNA处理、蛋白翻译、凋亡和转移,这与p53调节多种细胞途径的概念一致。一些已鉴定的蛋白质被鉴定为p53转录靶点或与p53相关,而其他蛋白质则没有与p53有关的报道。
eIF5A最初被描述为翻译起始因子(17),但对酵母的研究表明,它对一般蛋白质合成不是必需的(18). eIF5A与一般核输出受体CRM1相互作用并作为核质穿梭蛋白发挥作用(19). eIF5A的过度表达导致p53靶点的表达增加以及p53依赖性凋亡,表明它是p53的正调控因子(20). 我们在MMC处理24小时时观察到eIF5A的强p53依赖性上调,并在第一内含子中鉴定出p53结合基序(表1),表明eIF5A型是一个真正的p53靶基因。
hnRNP K蛋白是p53介导的细胞周期检查点基因转录所必需的(21). 它可以增强癌基因如c-myc和c-src的转录,并被认为可以促进细胞增殖、存活和迁移(22). 它还与染色质重塑、mRNA剪接、输出和翻译有关。我们鉴定了一个p53结合基序67-nt,位于hnRNP K公司发起人(表1). 因此,hnRNP K可能是p53的辅激活子,p53在正反馈回路中转录激活。有趣的是,我们观察到HCT116 wtp53中hnRNP K亚型a下调10倍+/+单元格(图3H(H)). 这可能是由于p53激活后hnRNP K亚型的稳定性降低或流动性改变,可能是翻译后修饰的结果。事实上,hnRNP K是磷酸化的体内(23),其与聚(C)的结合在磷酸化后被取消,表明磷酸化调节其活性(24).
同样,层粘连蛋白A/C以p53依赖性方式上调(图3C和4). 由LMNA公司基因属于A型层粘连蛋白,是核层粘连的主要成分。中的突变LMNA公司该基因与Hutchinson–Gilford早衰综合征相关,即早衰(25). 缺乏Zmptse24蛋白酶(导致前层蛋白A积聚)的小鼠表现出p53信号通路的激活(26). 过表达p53短亚型或产生p53激活羧基末端p53片段的小鼠显示出早期衰老的迹象(27,28). 这增加了层粘连蛋白A/C在某种程度上参与与p53相关的早衰表型的可能性。内含子1中p53结合位点的存在表明层粘连蛋白a/C是真正的p53靶点。
编号23-H1编码核苷二磷酸激酶(NDPK-A)的基因在野生型p53激活后mRNA和蛋白质水平上调(29). 它是细胞增殖的负调控因子,在转移性人类肿瘤(包括黑色素瘤和乳腺癌)中表达降低。我们观察到MMC处理的细胞中Nm23-H1的表达显著增加,这与p53相关,如Western blot分析和实时RT-PCR所证实的(图4和5天). 这个编号23-H1该基因包含一个假定的p53结合位点,位于转录起始位点上游5kb处(表1).
我们还将NPM鉴定为p53诱导蛋白(表1). NPM是一种丰富的核仁磷酸蛋白,参与核糖体蛋白的组装和转运,在细胞核和细胞质之间穿梭。NPM结合MDM2并阻止p53和MDM2之间的相互作用,从而调节p53(30). NPM还与p53结合,导致p53稳定性增加,应激时转录激活增加(31). 我们在转录起始位点上游2.4 kb处发现了一个p53结合位点。这表明NPM是p53诱导的蛋白靶点,与p53形成正反馈环。
hnRNP C1/C2蛋白是丰富的前RNA结合蛋白,参与前RNA成熟。观察到hnRNP C1/C2蛋白水平的p53依赖性诱导表明,p53可能调节这一过程。与eIF5A、hnRNP K、层粘连蛋白A/C和Nm23-H1相比,MMC治疗后hnRNP C1/C2 mRNA水平显著降低(图5E类). 因此,hnRNP C1/C2不是经典的p53转录靶点,而是转录后调控的靶点。可以想象,p53也以转录依赖的方式调节其他靶蛋白。这强调了蛋白质表达谱作为识别和表征下游p53靶点的工具的重要性,因为转录非依赖性靶点不会出现在微阵列分析中。有趣的是,2DE分析中的五个hnRNP C1/C2位点之一,这里称为hnRNP C1/C2 a,在p53中表达下调+/+单元格(图3我). 这可能是由于翻译后修饰,如磷酸化和/或小泛素样修饰物修饰。hnRNP C1/C2的磷酸化是其与premRNA结合所必需的,过度磷酸化导致premRNA释放hnRNP C1/C2。蛋白质磷酸酶2A(PP2A)随后的去磷酸化使蛋白质可用于下一轮磷酸化和mRNA处理(32). 我们发现PP2A在p53激活时下调(见下文)。这可能导致不能参与前RNA加工的高磷酸化hnRNP C1/C2蛋白的积累。值得注意的是,在不同刺激诱导的细胞凋亡过程中,hnRNP C1/C2蛋白被白细胞介素1β转化酶样蛋白酶和caspase 3裂解(33,34). 因此,p53诱导的凋亡可能以hnRNP C1/C2为靶点进行蛋白水解切割。值得注意的是,半胱天冬酶-3在MMC治疗8小时时被诱导(表1).
PP2A是一种具有多个细胞靶点的丝氨酸-苏氨酸磷酸酶。它具有由R5/B56调节亚单位特异性介导的抗凋亡活性。含有B56亚基的复合物的缺失导致细胞凋亡果蝇属(35). SV40小t抗原与PP2A结合并干扰其活性,有助于细胞转化(36). 此外,PP2A与多瘤病毒小t抗原(PyST)的复合物被证明可以抑制ARF介导的p53激活所需的信号(37). 在MMC治疗16小时后,我们检测到B56α-亚型受到2.3倍的抑制(表1). 这个PP2A型该基因在内含子1中有一个假定的p53结合位点,但其在p53介导的PP2A抑制中的作用尚不清楚。
根据我们的2DE分析,另一个确定的p53表达靶点Prx II下调,但Western blot分析没有发现任何显著的p53依赖性抑制。这表明p53下调Prx II的特定亚型和/或翻译后修饰形式,而不是整个库。Prx II调节细胞衰老和衰老。Prx II缺失小鼠胚胎成纤维细胞活性氧(ROS)水平升高,细胞衰老,PrxⅡ缺失小鼠皮肤老化加速(38). p53过度表达诱导活性氧的产生,导致线粒体成分的氧化降解,继而导致细胞凋亡(39). 我们的结果表明,p53介导的Prx II下调在ROS的生成和p53依赖性凋亡中起作用。这一观点与小鼠Prx V抑制p53诱导的ROS生成和凋亡的观察结果一致(40). 我们没有观察到Prx II在mRNA水平上受到任何抑制,这表明该基因不是p53的直接转录靶点。
氨酰-tRNA合成酶在蛋白质合成中起主要作用,催化特定氨基酸与其同源tRNA的连接,但也可以参与多种细胞过程,如RNA运输、rRNA合成、凋亡、血管生成和炎症。在人类细胞中,由于选择性剪接,TrpRS表达为全长的主要形式和被截断的形式,即mini-TrpRS。Mini-TrpRS被证明抑制视网膜血管生成,而全长TrpRS则被认为主要在翻译中起作用(41). 我们观察到在MMC治疗24小时时,p53的全长TrpRS(MW 53 kDa)下调2.7倍+/+单元格(表1). Western blot分析证实了这一点,而实时PCR未发现任何变化(图4和5G公司)表明p53独立于转录下调TrpRS。p53可能诱导TrpRS的翻译后修饰,导致任一蛋白变体的下调。有证据表明,p53可能通过下调TrpRS来抑制翻译,这一点以前没有报道过。
我们对p53依赖性蛋白质组的分析已经鉴定出55种可能的p53调节蛋白,其中许多是未知的p53靶点。这些蛋白质分为多个类别。根据我们的发现,我们可以将p53与mRNA处理(hnRNP C1/C2)、翻译(TrpRS)、氧化还原调节(Prx II,硫氧还蛋白)、凋亡(caspase-3,eIF5A)、细胞生长控制(14-3-3σ,Nm23-H1)、衰老(层粘连蛋白A/C,PrxⅡ)、Ras信号和MAP激酶途径(GRB2,MAPK8)、蛋白酶体降解(TBP-1)、,磷酸酶信号(PP1和PP2A)和伴侣活性(Hsp27,Hsp60)。进一步的工作应该确认已鉴定蛋白质的p53依赖性调节,并在转录水平上检查p53依赖的调节。
我们的研究结果支持这样一种观点,即p53肿瘤抑制剂通过多种相互连接的途径以及转录依赖性和转录非依赖性机制,在多个水平上调节细胞生长、存活和其他细胞过程。本研究还表明,全局蛋白质组分析适合于鉴定新的p53靶点,并表明p53参与了许多蛋白质的翻译后修饰。进一步分析p53调节蛋白可以更好地了解p53介导的肿瘤抑制背后的分子机制。这一分析也可能导致确定新的靶点,以改进癌症治疗。
材料和方法
细胞和细胞培养。
HCT116 p53型+/+和p53−/−结肠癌细胞按上述方法生长(4). 以10μg/ml的浓度添加MMC(西格玛,密苏里州圣路易斯)以激活内源性p53。添加Staurosporine(100 nM)(Sigma)诱导p53诱导的依赖性细胞死亡。细胞凋亡评估如下(4). 细胞在MMC处理前用30μM ZVAD(MP Biomedicals,Aurora,OH)处理2 h,以抑制半胱天冬酶。用CaspaTag-Pan半胱天冬酶试剂盒(Chemicon,Temecula,CA)通过流式细胞术测量半胱天冬酶活性。
Western Blot分析和免疫染色。
根据标准程序,使用以下一级抗体进行Western blot分析:来自Santa Cruz Biotechnology(加州圣克鲁斯)的p53单克隆和MDM2多克隆;来自Novocastra(英国纽卡斯尔)的hTERT单克隆抗体;来自Pharmingen(加利福尼亚州圣地亚哥)的p21、lamin A/C和eIF5A单克隆;来自Sigma的β-actin单克隆抗体;以及来自Abcam(马萨诸塞州剑桥)的hnRNP C1/C2、Nm23-H1和Prx II单克隆和hnRNP K多克隆。按照描述进行免疫荧光染色(4).
实时PCR。
根据制造商的说明,使用逆转录试剂盒(Invitrogen,Carlsbad,CA)对总RNA进行逆转录。使用ABI Prism 7500仪器(加利福尼亚州福斯特市应用生物系统公司)进行实时PCR。
2D凝胶电泳。
如前所述进行等电聚焦(IEF)和多肽分离(SDS/PAGE)(42). 对于第一维度,每个固定化pH梯度(IPG)条带装载250–800μg蛋白质,并在约60000 volt-h的蛋白质IEF细胞(Bio-Rad)中聚焦。对于第二个维度,浇注10–13%线性SDS/PAGE梯度凝胶。在SDS/PAGE凝胶顶部应用IPG条带,并在42000伏特-小时的条件下进行20小时的电泳。为了进行图像分析,分析凝胶用硝酸银染色。使用Sypro-Ruby染色溶液(Molecular Probes,Eugene,OR)观察制备凝胶上的斑点。有关更多信息,请参阅SI文本.
扫描和图像分析。
按照概述进行扫描(42). 构建了四个匹配集(每个pH值范围一个)。来自不同时间点的凝胶中的蛋白质斑点与参考凝胶中的斑点相匹配。将斑点强度归一化,并确定相对染色强度。通过Student’st吨和Mann-Whitney测试(P(P)< 0.05).
凝胶内消化和质谱法。
使用EXQuest斑点切割器(Bio-Rad)去除2DE凝胶中的Sypro-Ruby蛋白斑点。按照说明进行凝胶内手动消化(43). 在Ultraflex TOF/TOF仪器(Bruker Daltonics,Billerica,MA)中进行肽质量指纹分析。使用ProFound进行蛋白质身份搜索(http://prowl.rockefeller.edu/profound_bin/). 通过分析自溶胰蛋白酶裂解产物实现内部校准,精确度为±0.05 Da。根据测量结果判断显著性z(z)-数值、匹配肽质量的数量以及蛋白质的实验和理论物理性质之间的一致性。
致谢
我们感谢Bert Vogelstein(马里兰州巴尔的摩约翰霍普金斯大学)提供的HCT116细胞和Thierry Soussi提供的宝贵意见。这项工作得到了瑞典癌症协会(Cancerfonden)和卡罗林斯卡研究所(Karolinska Institutet)的支持。
缩写
- 二维工程
2D凝胶电泳
- 多媒体卡
丝裂霉素C
- ROS公司
活性氧物种。
工具书类
-
1Vousden KH,Lu X.Nat Rev癌症。2002;2:594–604. doi:10.1038/nrc864。[内政部] [公共医学] [谷歌学者]
-
2Wang Y、Szekely L、Okan I、Klein G、Wiman KG。癌基因。1993;8:3427–3431.[公共医学] [谷歌学者]
-
三。Prisco M、Hongo A、Rizzo MG、Sacchi A、Baserga R.Mol Cell Biol。1997;17:1084–1092. doi:10.128/mcb17.3.1084。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
4Rahman R、Latonen L、Wiman KG。癌基因。2005;24:1320–1327. doi:10.1038/sj.onc.1208232。[内政部] [公共医学] [谷歌学者]
-
5Caelles C,Helmberg A,Karin M.自然。1994;370:220–223. doi:10.1038/370220a0。[内政部] [公共医学] [谷歌学者]
-
6Mihara M、Erster S、Zaika A、Petrenko O、Chittenden T、Pancoska P、Moll UM、Mol Cell。2003;11:577–590. doi:10.1016/s1097-2765(03)00050-9。[内政部] [公共医学] [谷歌学者]
-
7Donehower LA、Harvey M、Slagle BL、McArthur MJ、Montgomery CA,Jr、Butel JS、Bradley A.Nature。1992;356:215–221. doi:10.1038/356215a0。[内政部] [公共医学] [谷歌学者]
-
8Symonds H、Krall L、Remington L、Saenz-Robles M、Lowe S、Jacks T、Van Dyke T.Cell。1994;78:703–711. doi:10.1016/0092-8674(94)90534-7。[内政部] [公共医学] [谷歌学者]
-
9Attardi LD、Lowe SW、Brugarolas J、Jacks T.EMBO J,1996年;15:3693–3701.[PMC免费文章] [公共医学] [谷歌学者]
-
10Kannan K、Amariglio N、Rechavi G、Jakob-Hirsch J、Kela I、Kaminski N、Getz G、Domany E、Givol D.癌基因。2001;20:2225–2234. doi:10.1038/sj.onc.1204319。[内政部] [公共医学] [谷歌学者]
-
11Zhao R、Gish K、Murphy M、Yin Y、Notterman D、Hoffman WH、Tom E、Mack DH、Levine AJ。基因开发2000;14:981–993。[PMC免费文章] [公共医学] [谷歌学者]
-
12Mirza A、Wu Q、Wang L、McClanahan T、Bishop WR、Gheyas F、Ding W、Hutchins B、Hockenberry T、Kirschmeier P等。癌基因。2003;22:3645–3654. doi:10.1038/sj.onc.1206477。[内政部] [公共医学] [谷歌学者]
-
13Wei CL、Wu Q、Vega VB、Chiu KP、Ng P、Zhang T、Shahab A、Yong HC、Fu Y、Weng Z等。Cell。2006;124:207–219. doi:10.1016/j.cell.2005.10.043。[内政部] [公共医学] [谷歌学者]
-
14Wang L,Wu Q,Qiu P,Mirza A,McGuirk M,Kirschmeier P,Greene JR,Wang Y,Pickett CB,Liu S.J生物化学。2001;276:43604–43610. doi:10.1074/jbc。M10657200。[内政部] [公共医学] [谷歌学者]
-
15Bunz F、Dutriaux A、Lengauer C、Waldman T、Zhou S、Brown JP、Sedivy JM、Kinzler KW、Vogelstein B.科学。1998;282:1497–1501。doi:10.1126/science.282.5393.1497。[内政部] [公共医学] [谷歌学者]
-
16Hoh J、Jin S、Parrado T、Edington J、Levine AJ、Ott J.美国国家科学院院刊2002;99:8467–8472. doi:10.1073/pnas.132268899。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
17好时JW。生物化学年度收益。1991;60:717–755. doi:10.1146/annrev.bi.60.0071191.003441。[内政部] [公共医学] [谷歌学者]
-
18Kang HA,好时JW。生物化学杂志。1994;269:3934–3940.[公共医学] [谷歌学者]
-
19Rosorius O、Reichart B、Kratzer F、Heger P、Dabauvalle MC、Hauber J.细胞科学杂志。1999年;112(第14部分):2369-80。doi:10.1242/jcs.112.14.2369。[内政部] [公共医学] [谷歌学者]
-
20李亚龙,李海燕,金碧芬,叶群恩,周涛,于晓东,潘X,曼建华,何凯,于敏,等.生物化学杂志。2004;279:49251–49258. doi:10.1074/jbc。M407165200。[内政部] [公共医学] [谷歌学者]
-
21Moumen A、Masterson P、O'Connor MJ、Jackson SP.Cell。2005;123:1065–1078. doi:10.1016/j.cell.2005.09.032。[内政部] [公共医学] [谷歌学者]
-
22Bomsztyk K、Denisenko O、Ostrowski J.生物论文。2004;26:629–638. doi:10.1002/bies.20048。[内政部] [公共医学] [谷歌学者]
-
23Schulery DS、Ostrowski J、Denisenko ON、Stempka L、Shnyreva M、Suzuki H、Gschwendt M、Bomsztyk K.生物化学杂志。1999年;274:15101–15109. doi:10.1074/jbc.274.21.15101。[内政部] [公共医学] [谷歌学者]
-
24Dejgaard K、Leffers H、Rasmussen HH、Madsen P、Kruse TA、Gesser B、Nielsen H、Celis JE.分子生物学杂志。1994;236:33–48. doi:10.1006/jmbi.1994.1116。[内政部] [公共医学] [谷歌学者]
-
25Goldman RD、Shumaker DK、Erdos MR、Eriksson M、Goldman AE、Gordon LB、Gruenbaum Y、Khuon S、Mendez M、Varga R、Collins FS。美国国家科学院院刊2004;101:8963–8968. doi:10.1073/pnas.0402943101。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
26Varela I、Cadinaos J、Pendas AM、Gutierrez Fernandez A、Folgueras AR、Sanchez LM、Zhou Z、Rodriguez FJ、Stewart CL、Vega JA等。《自然》。2005;437:564–568. doi:10.1038/nature04019。[内政部] [公共医学] [谷歌学者]
-
27Maier B、Gluba W、Bernier B、Turner T、Mohammad K、Guise T、Sutherland A、Thorner M、Scrable H.Genes Dev.2004;18:306–319. doi:10.1101/gad.1162404。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
28Tyner SD、Venkatachalam S、Choi J、Jones S、Ghebranious N、Igelmann H、Lu X、Soron G、Cooper B、Brayton C等人《自然》。2002;415:45–53. doi:10.1038/415045a。[内政部] [公共医学] [谷歌学者]
-
29Chen SL,Wu YS,Shieh HY,Yen CC,Shen JJ,Lin KH。Mol Carcinog。陈士林,吴爱思,谢慧,甄聪,沈俊杰,林坤。2003;36:204–214. doi:10.1002/mc.10110。[内政部] [公共医学] [谷歌学者]
-
30Kurki S、Peltonen K、Latonen L、Kiviharju TM、Ojala PM、Meek D、Laiho M.癌细胞。2004;5:465–475。doi:10.1016/s1535-6108(04)00110-2。[内政部] [公共医学] [谷歌学者]
-
31科伦坡E、Marine JC、Danovi D、Falini B、Pelicci PG.Nat Cell Biol。2002;4:529–533. doi:10.1038/ncb814。[内政部] [公共医学] [谷歌学者]
-
32Mayrand SH、Dwen P、Pederson T.美国国家科学院院刊1993;90:7764–7768. doi:10.1073/pnas.90.16.7764。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
33Waterhouse N、Kumar S、Song Q、Strike P、Sparrow L、Dreyfuss G、Alnemri ES、Litwack G、Lavin M、Watters D.J Biol Chem。1996;271:29335–29341. doi:10.1074/jbc.271.46.29335。[内政部] [公共医学] [谷歌学者]
-
34Brockstedt E、Rickers A、Kostka S、Laubersheimer A、Dorken B、Wittmann-Liebold B、Bommert K、Otto A.生物化学杂志。1998;273:28057–28064. doi:10.1074/jbc.273.43.28057。[内政部] [公共医学] [谷歌学者]
-
35Silverstein AM,Barrow CA,Davis AJ,Mumby MC,美国国家科学院院刊,2002年;99:4221–4226. doi:10.1073/pnas.072071699。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
36阿罗约·JD,哈恩·WC。致癌物。2005;24:7746–7755. doi:10.1038/sj.onc.1209038。[内政部] [公共医学] [谷歌学者]
-
37Moule MG、Collins CH、McCormick F、Fried M.Proc Natl Acad Sci USA,2004年;101:14063–14066。doi:10.1073/pnas.0405533101。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
38Han YH、Kim HS、Kim JM、Kim-SK、Yu DY、Moon EY。FEBS信函。2005;579:4897–4902. doi:10.1016/j.febslet.2005.07.049。[内政部] [公共医学] [谷歌学者]
-
39Polyak K、Xia Y、Zweier JL、Kinzler KW、Vogelstein B.Nature。1997;389:300–305. doi:10.1038/38525。[内政部] [公共医学] [谷歌学者]
-
40Zhou Y、Kok KH、Chun AC、Wong CM、Wu HW、Lin MC、Fung PC、Kung H、Jin DY。生物化学生物物理研究委员会。2000;268:921–927. doi:10.1006/bbrc.2000.2231。[内政部] [公共医学] [谷歌学者]
-
41Wakasugi K、Slike BM、Hood J、Otani A、Ewalt KL、Friedlander M、Cheresh DA、Schimmel P.美国国家科学院院刊,2002年;99:173–177. doi:10.1073/pnas.012602099。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
42Roblick UJ、Hirschberg D、Habermann JK、Palmberg C、Becker S、Kruger S、Gustafsson M、Bruch HP、Franzen B、Ried T等。细胞分子生命科学。2004;61:1246–1255. doi:10.1007/s00018-004-4049-4。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
43Hellman U,Bhikhabhai R.快速通讯质谱。2002;16:1851–1859。doi:10.1002/rcm.805。[内政部] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。


{kind=link}
{kind=link}
{kind=link}