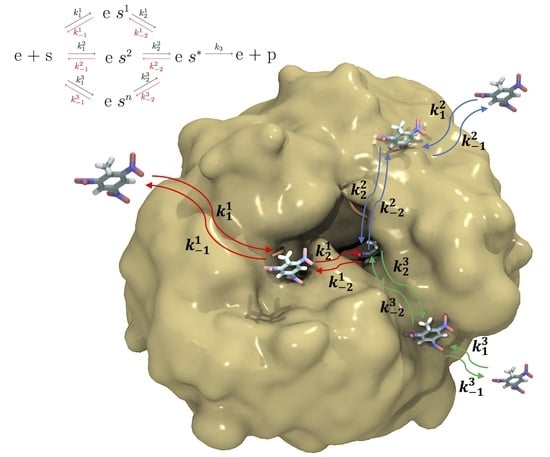
图1。动力学方案:(一)显示了不考虑PCBS和(b条)显示了包含PCBS的动力学方案。K是一个基本步骤的动力学常数,其中正下标表示产品形成步骤,负下标表示反向过程。对于方案(b条),其中包括n个PCBS,上标使每个PCBS个性化。
图2。催化效率随PCBS数量的增加而增加。对MM模型的6个动力学常数条件(第一点i=0)和i=1、2、3、4、5和5 PCBS进行了数值模拟。对于每个模拟,催化效率()绘制为PCBS数量的函数。由于曲线b和d以及e和f重叠,因此可以用分段线观察到这些曲线。
表1。酶动力学的数值模拟.对于模拟,底物结合的动力学常数,离解常数和催化常数酶浓度为0.0002 mM,底物浓度为0.1、0.2、0.5、5、10、100 mM。为了分析动力学,取每个底物浓度的前10个点的斜率,从倒数加倍中获得动力学参数。
| 动力学参数 | Michaelis–Menten公司* | | | | | |
|---|
| | | | | | |
| | | | | | |
| 催化效率 | | | | | | |
表2。酶动力学的数值模拟.对于模拟,底物结合的动力学常数,离解常数和催化常数酶浓度为0.0002 mM,底物浓度为0.1、0.2、0.5、5、10、100 mM。为了进行动力学分析,取每个底物浓度的前10个点的斜率,从倒数加倍中获得动力学参数。
| 动力学参数 | Michaelis–Menten公司* | | | | | |
|---|
| | | | | | |
| | | | | | |
| 催化效率 | | | | | | |
表3。酶动力学的数值模拟.对于模拟,底物结合的动力学常数,,离解常数和催化常数酶浓度为0.0002 mM,底物浓度为0.1、0.2、0.5、5、10、100 mM。为了进行动力学分析,取每个底物浓度的前10个点的斜率,从倒数加倍中获得动力学参数。
表3。酶动力学的数值模拟.对于模拟,底物结合的动力学常数,,离解常数和催化常数酶浓度为0.0002 mM,底物浓度为0.1、0.2、0.5、5、10、100 mM。为了进行动力学分析,取每个底物浓度的前10个点的斜率,从倒数加倍中获得动力学参数。
| 动力学参数 | Michaelis–Menten公司* | | | | | |
|---|
| | | | | | |
| | | | | | |
| 催化效率 | | | | | | |







{kind=link}
{kind=link}
{kind=link}
{kind=link}