摘要
在西方社会,与年龄相关的黄斑病变(AMD)占盲人登记的50%以上。AMD患者分为早期疾病(视力保持良好)和晚期疾病(中心视力丧失)。直到最近,还没有可用的治疗方法来改变病程。现在,最常见的晚期AMD形式-脉络膜新生血管-对抗VEGF治疗有反应;尽管在这些病例中有一部分视力下降得到了改善,但没有任何治疗方法可以改变疾病从早期到晚期的演变。然而,正如本综述所讨论的,过去几年的研究表明AMD的几个特征可能适合治疗。描述了治疗的潜在靶点,并讨论了可能的治疗方法。
老年性黄斑病变(AMD)是一种常见的眼部疾病,其特征是视网膜受损导致视野中心失明。它通常影响50岁以上的成年人。重要的是,AMD在西方社会被公认为导致50%以上的盲人登记,现已被指定为世界上主要致盲疾病之一(1–4).与目前AMD的高发病率相比,在19世纪,AMD被认为是一种罕见的疾病,被描述为“老年人脉络膜视网膜疾病”(5).有证据表明,目前AMD发病率的增加不能完全用预期寿命的增加来解释(6).此外,这种疾病现在被认为是30年前罕见的社会的负担。过去有充分的证据表明,AMD在一些非欧洲血统人群中的发病率较低(7,8).然而,在过去20年里,这种紊乱在日本的城市社区已经很常见(9–12)在过去3年中,医院转诊率翻了一番(M.Uyama,个人通信)。出版物表明,东亚其他地方的流行率也可能发生类似的变化(13–15).格陵兰岛的老年因纽特人黄斑病的发病率也高得惊人(16,17).
多年来,家族性AMD的发病已被公认,并且最近已确定许多基因具有患AMD的风险,包括补体因子H(CFH公司),循环流化床,补足成分2(指挥与控制),C3类和年龄相关性黄斑病变易感性2/HtrA丝氨酸肽酶1(ARMS2系统/HTRA1型)10q26基因座(18–25).由于大多数基因编码与补体级联有关的蛋白质,补体级联有助于先天免疫系统清除病原体,因此先天免疫系统的失调可能在AMD的发病机制中起重要作用。CFH尤其令人感兴趣。在50%的AMD患者中发现CFH中产生Y402H CFH变体的一个多态性,并预测该多态性会导致补体级联反应的放松(18–25).防护CFH公司单倍型也存在(21,26).此外CFH公司导致2型系膜毛细血管肾病,这是一种以肾小球基底膜增厚和模拟AMD的眼底(晶状体对面的眼睛内表面)改变为特征的疾病(27).除了AMD风险的遗传因素外,环境因素也在疾病发展中起作用(28).有大量证据表明,吸烟和肥胖都会带来风险,而且它们对基因的影响是相加的(29).
参与疾病过程的眼部结构是感光细胞,即视网膜外侧负责将光子转换为动作电位的特殊神经元;视网膜色素上皮(RPE),为感光细胞提供代谢支持的极性细胞单层;Bruch膜,介于RPE和脉络膜之间的胶原和弹性层的复合物;脉络膜毛细血管,内脉络膜中的毛细血管床,提供外视网膜的代谢需求(图1).这些结构在新陈代谢上相互依赖,在布鲁赫膜上有相当大的新陈代谢交换。感光细胞的外段由一堆膜组成,这些膜形成圆盘,其中含有色素,可吸收光子并启动视觉过程。感光细胞由两种类型组成:视锥细胞在强光下调节视觉(即明视),视杆细胞在暗光下调节视力(即暗视)。每天,每个感光细胞外段的远端被RPE脱落和吞噬。吞噬体与溶酶体合并形成吞噬溶酶体。溶酶体酶降解吞噬体中的物质,其中一些物质被再循环回感光细胞,形成新的外节圆盘。据信,剩余的降解物质向外排入布鲁赫膜,由脉络膜毛细血管清除。RPE中未降解的物质形成电子密度高的残体,随着年龄的增长,残体积聚在RPE中。残余体的一个重要组成部分由全经视网膜和乙醇胺(A2-E)的组合组成,具有自发荧光,并且具有高度的抗降解性。这种荧光团的形成始于外段。据认为,全跨视网膜在漂白后从色素释放到椎间盘间隙,并在与膜脂磷脂酰乙醇胺结合后转移到细胞质间隙。如果2个全跨视网膜分子与膜脂结合,则复合物不会翻转。随着椎间盘年龄的增长,视网膜/乙醇胺混合物的数量也在增加。该化合物在吞噬过程中被带入RPE细胞,然后在吞噬体的酸性环境中转化为脂褐素。这些过程在AMD中变得不受调节,这些组织在整个眼睛中都会发生变化,尽管它们在黄斑区最为明显,黄斑区为中心视力的分支,包含高密度的视锥细胞。
图1。与AMD相关的眼部解剖。

(A类)眼睛的横切面,显示眼睛内侧的视网膜。 (B类)视网膜的横切面,显示神经视网膜及其外部RPE和脉络膜。INL,内核层;ONL,外核层。 (C类和D类)图表(C类)和光微观视图(D类)参与AMD的视网膜组织。RPE的顶端微绒毛与感光器外节的远端部分交叉。视网膜色素上皮外是脉络膜,其间插入布鲁赫膜。
AMD在临床上可分为早期和晚期。在早期疾病中,症状很少,视力正常。然而,在眼底,布鲁赫膜中可以看到称为“酒石”的局部沉积物。它们的分布和大小因患者而异,尽管这些属性在个体眼睛之间高度一致。RPE也可能有不规则的色素沉着。
晚期AMD的3种类型导致中心视力丧失。最常见的形式是脉络膜新生血管(CNV),当脉络膜毛细血管的血管向内或穿过Bruch膜时发生(图2、A和B)。视网膜色素上皮脱离(PED),液体积聚在RPE和Bruch膜之间(图2C) ,相对来说是不常见的。在地理萎缩(GA)中,RPE和感光细胞有明确的丢失。GA通常被认为是疾病过程的默认途径,而PED和CNV在其进化过程中作为反应性事件发生。CNV有成功的治疗方法,但PED或GA没有经证实的治疗方法可用,也没有办法控制早期疾病中发生的事件。在这篇综述中,我试图确定新治疗策略可能基于的治疗目标。由于AMD是一种多因素疾病,其危险因素在社区中很常见,因此很难定性区分与年龄相关的变化和AMD,并且差异可能仅在于变化的严重程度。因此,许多研究仅集中于年龄相关的变化,假设这些发现将进一步加深对AMD的理解。
图2。AMD的渗出性病变。

(A类和B类)在CNV中,血管向内向脉络膜生长,并可能在Bruch膜内增殖(A类)或越过RPE在视网膜下腔生长(B类). (C类)在RPE脱离过程中,脂质积聚在Bruch膜中,使其具有疏水性。RPE将水向外移向脉络膜(箭头);由于无法自由向外通过,水导致RPE从Bruch膜上脱落。
AMD患者脉络膜的结构变化
由于所检查的标本较少,因此有关AMD患者脉络膜毛细血管组织病理学变化的文献相对较少。在健康的年轻人中,脉络膜毛细血管由正弦复合体构成,有窗,缺乏紧密连接。据信,脉络膜毛细血管的发育部分取决于RPE向外构成的VEGF表达(30–34).
一项形态计量学研究发现,无AMD患者的脉络膜毛细血管密度随年龄增长而降低(35).在另一项研究中,使用氯丁橡胶铸型来显示从正弦系统到管状血管系统的变化(图三和参考。36).在晚期AMD中,脉络膜毛细血管会消失和变窄(37–40).
图3。脉络膜铸型内表面的扫描电子图像。

(A类)年轻供体的脉络膜毛细血管是一个密集的正弦网络,看不到外脉络膜的大血管。 (B类)在87岁的受试者中,毛细血管是管状的,毛细血管之间有很大的间隙。图由J.Olver复制。
索斯比眼底营养不良症是一种单基因黄斑变性疾病,在生命的第三或第四个十年中会导致中心视力丧失;它与AMD有许多共同的病理特征(41).其特征是Bruch膜主要增厚,荧光素血管造影显示脉络膜充盈期延长(42).通常,静脉注射荧光素钠时,染料会从脉络膜毛细血管中自由泄漏,并与布鲁赫膜中的极性化合物结合。拍照时,在染料进入眼睛循环的1秒内,这会产生均匀的背景荧光。在Sorsby眼底营养不良症中,背景荧光可能需要10秒以上才能持续。这种延迟可能是由于窗孔的丢失以及毛细床从正弦状态变为管状状态所致。据认为,弥漫性加厚的Bruch膜是VEGF向脉络膜扩散的屏障,导致毛细血管床发生变化(43,44).这种血管造影异常也在早期AMD患者中发现(45).这一临床观察的潜在意义在于,视网膜的离散区域在低光条件下(即暗视)的视觉阈值升高高达3.4 log单位,以及缓慢的暗适应(患者有症状),与脉络膜异常灌注区域密切相关(46,47).视力丧失不太明显。
AMD患者脉络膜的改变很可能是对邻近组织改变的反应,尽管有人提出了另一种选择,即可能存在内在改变(48).由此引起的外视网膜代谢供应的减少可能对疾病的发生起重要作用。脉络膜目前尚未被视为治疗的良好靶点。
AMD患者Bruch膜的结构变化
通过电子显微镜和光学显微镜已经确定了老化与布鲁赫膜厚度之间的直接关系(49,50).然而,在一项研究中,比较布鲁赫膜厚度与年龄,发现老年人在电子显微镜和光学显微镜上都有很大差异(51).因此,大约一半的厚度变化必须由年龄以外的因素来解释,如遗传或环境影响。增厚被认为是由RPE向外排放的废料清理不彻底引起的,至少部分是这样,从而导致沉积物堆积。
已经对矿床的性质进行了几项研究。随着对PED发病机制的讨论,有关含量及其对增厚的Bruch膜功能的潜在影响的线索也随之出现。离子和水不断向外运动,从视网膜外侧向脉络膜方向运动,据推测,Bruch膜的导水性降低会阻碍水向脉络膜的运动,导致水积聚在视网膜色素上皮下间隙(图2C和参考。52).这一概念要求Bruch膜含有高脂含量,这将增加流体流动阻力。为了验证这些假设,我们进行了一系列调查,并从组织病理学、生物化学、生物物理和临床观察中获得了支持(53–59).一项使用组织化学染色对1至95岁的人的捐赠者眼睛进行的冷冻组织研究表明,随着年龄的增长,老年人体内脂质的数量和形式都有很大差异(图4和参考。53).一些眼睛只染中性脂质,一些眼睛主要染磷脂,其他眼睛中性脂质和磷脂染色相同。为了证实这些观察结果,用薄层色谱法和气相色谱法分析了用脂类溶剂从新鲜人眼组织中提取的物质(54,55).分离后,通过质谱鉴定化学物种。本研究证实,Bruch膜中总脂质的数量随着年龄的增长而增加。从50岁以下捐赠者的标本中提取的脂质很少或没有;在50岁以上捐赠者的标本中,增长呈指数级。60岁以上捐赠者的眼睛显示,从相似年龄的捐赠者身上提取的总脂质有很大差异,并且磷脂与中性脂肪的比率在相同年龄的捐献者身上不同。中性脂质与磷脂的比率与脂质总量无关。发现主要的脂质种类是磷脂和脂肪酸,而不是胆固醇和胆固醇酯,并且只有50%的磷脂是磷脂酰胆碱,这一发现导致了这样的结论:脂质是细胞来源的(可能是RPE),而不是血浆来源的(55).在这方面,脂质沉积不同于来自血浆的动脉粥样硬化斑块和老年弧中的脂质沉积。因此,AMD与高脂血症之间的关系并不一定是可以预料的,而降脂药物可能是没有帮助的(56).Curcio及其同事使用不同的提取方法发现,胆固醇和胆固醇酯是Bruch膜中的主要脂质,而不是磷脂(57).然而,与之前的研究一样(55)根据胆固醇的性质,可以得出结论,这些脂质来源于RPE(57).最后,对Bruch膜的水力传导率的测量表明,随着年龄的增长,水力传导率降低(58,59)50岁以后,液体流动阻力与脂肪含量之间存在密切的直线关系。因此,有充分证据表明,布鲁赫膜的生物物理特性随着老化而改变。在极端情况下,这些变化很可能会导致PED,但它们可能对老化的视网膜产生深远影响,并在所有形式的AMD中发挥重要作用。
图4。眼睛冰冻部分的脂质染色。
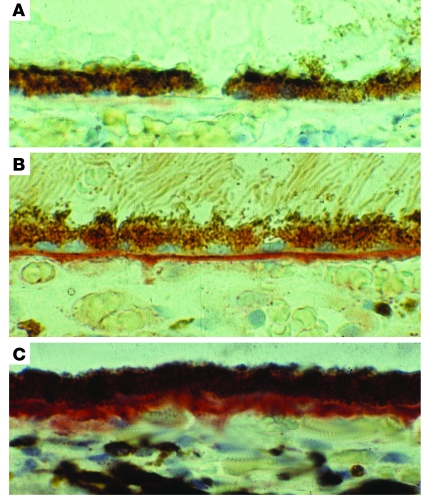
16岁儿童冷冻视网膜横截面的显微照片(A类),44岁(B类),84岁(C类)供体用油红O染色,表明Bruch膜中的脂质量随年龄增长而增加。
临床观察旨在支持生化含量和布鲁赫膜厚度影响后续临床行为的概念。据推测,荧光素血管造影可以显示局灶性Bruch膜沉积的脂质含量,这可能反映了弥漫性脂质沉积的存在或不存在。有人认为,亲水性的核糖将允许水溶性荧光素钠自由扩散到异常沉积物中,并且染料将与极性分子结合,从而使它们在荧光素血管造影中产生高荧光。相反,如果浮肿荧光减弱,则表明它们是疏水性的,因为存在中性脂质。这一结论得到组织学观察的支持,其中显示荧光素钠的体外结合与酒石的生化含量密切相关(60).富含中性脂质的酒不与荧光素结合,而脂质含量低的酒则与荧光素强结合。
Bruch膜对水流的最大阻力预计出现在注定要遭受PED撕裂的眼睛中,其中分离组织中的切向应力足以导致其破裂。确定一只眼睛的RPE撕裂导致另一只眼睛发生类似事件的高风险(61)提供了在临床环境中测试该概念的机会。血管造影术中,比较了泪液中有酒渣鼻的对眼和视网膜下新生血管的对眼的酒渣鼻荧光;前者的血管造影显示,与后者相比,丘疹更大,更融合,荧光更少(62).因此,临床观察支持脂质对Bruch膜的生物物理特性有重大影响并有助于AMD的一般概念。
有大量的脂质通过Bruch膜进行转运,脂质被认为会积聚,因为它们无法自由通过加厚的Bruch薄膜。这要求Bruch膜变厚,作为这种脂质积聚的要求。已经对老化的Bruch膜中的蛋白质进行了分析,因为这可能会导致膜增厚。最近的研究表明,一些与免疫系统相关的蛋白质,如C3、C5b–C9和CFH,在AMD的Bruch膜中大量存在(63).这些观察结果强调了AMD患者免疫系统紊乱的潜在重要性。然而,与身体其他部位的炎症不同,没有炎症细胞浸润布鲁赫膜。β-淀粉样蛋白也已被鉴定(64–67).在布鲁赫膜的内部,有高水平的卵黄凝集素,可能对免疫攻击起保护作用(68,69).蛋白质的来源尚不清楚,因为一些成分有RPE表达,而主要的贡献可能来自血浆(70).蛋白质的状态尚不清楚,但间接证据表明,包括CFH在内的一些蛋白质是寡聚的,这可能是由高水平的锌或其他金属离子诱导的(71,72).在Bruch膜中,锌的含量非常高,生物可利用锌的含量是导致体外CFH齐聚所需锌含量的数倍(71).与AMD高风险相关的CFH变异体Y402H预计比与低风险疾病相关的变异体更容易寡聚,因为氨基酸变化提供了额外的锌结合位点。因此,积聚在Bruch膜中的至少一些蛋白质可能不具有单体的生物特性,因此可能不会通过其炎症作用影响眼睛功能(73,74).相反,由于它们的累积体积,它们可能会成为新陈代谢交换的屏障。
对Bruch膜中物质积累的可能机制的进一步了解来自于Cfh(立方英尺)–/–鼠标(75).基因敲除不一定产生与多态性产生的表型同源的表型,因为蛋白质的缺失不同于修饰蛋白质功能的氨基酸变化。此外,小鼠的免疫系统与人类的不同,而且小鼠没有黄斑。然而,如果CFH活性的降低很重要,那么这些观察可能有助于理解AMD。在Cfh(立方英尺)–/–小鼠肾小球基底膜增厚,但令人惊讶的是,Bruch膜比年龄匹配的小鼠薄。这表明,仅免疫系统的失调可能无法解释布鲁赫膜的增厚,CFH蛋白的存在对这一过程很重要。
因此,有大量证据表明,Bruch膜增厚导致代谢交换阻抗和液体运动,这可能在AMD的发病机制中很重要。可以考虑几种治疗方法。通过长期使用抗炎药降低组分蛋白质的可用性可能会减缓疾病进程。一旦增厚形成,可以通过使用抗体或锌缓冲液来分解低聚物,正如阿尔茨海默病所建议的那样(76).然而,快速生成单体存在潜在风险,例如对RPE的补体攻击(73).或者,可以动员脂质。所有这些方法都会增加视网膜色素上皮和脉络膜毛细血管之间的导水率和代谢交换。此外,导水率的增加会使视网膜色素上皮表达的血管内皮生长因子扩散,并导致脉络膜循环和窗孔密度增加。
AMD患者RPE的结构变化
荧光残体的积累可作为RPE中老化的指标。年龄与自体荧光和残留体数量之间存在二次关系,分别通过光学显微镜和电子显微镜的自体荧光成像进行测量(51).老年人中每一种光感受器的积累速度减慢并不奇怪,因为老年人的感光细胞数量减少(77).然而,鉴于在老年人中观察到的广泛差异,年龄与自体荧光之间的关系并不密切(34).得出的结论是,50%的自体荧光和残留体的变化不能用老化来解释,怀疑是遗传或环境因素在决定变化中起作用。最令人惊讶的是自体荧光和残余体积之间的微弱关系(51).这是直接的,这是意料之中的,因为自体荧光是从残留体中衍生出来的。回顾过去,标本之间的差异不应令人惊讶,因为残留体内只有一小部分物质会发出荧光,而这一比例可能会受到饮食中维生素a含量等因素的影响(56).与此相一致的是,如果啮齿动物的饮食中维生素a含量低,残留的身体就不会发出荧光(78).在饮食中富含维生素a的室友中,RPE的残余体内含量相似,但它们发出明亮的荧光。从这一观察结果可以得出结论,那些自身荧光水平高的患者饮食中维生素a含量高。或者,他们可能有遗传倾向,例如ATP-结合盒、亚家族a、成员4中的变异所导致的遗传倾向(ABCA4公司)基因。这种变异会增加光感受器外段的脂褐素含量,从而增加RPE,如Stargard病(由ABCA4突变引起)和阿布卡4-空鼠标(79,80).饮食维生素A的重要性得到了雷克雅未克眼科研究的进一步支持,在冰岛,GA占AMD失明病例的80%,而英国约占25%(81).由于饮食中含有大量鱼类,以及服用维生素A补充剂的习惯做法,该社区的膳食维生素A摄入量非常高。正如所料,奥斯陆GA的流行程度介于雷克雅未克和伦敦之间(82).
这些发现的临床相关性通过现在在体内成像RPE自身荧光的能力得到了强调(83).这是通过使用共聚焦扫描激光检眼镜(SLO)实现的,其激发波长为488nm,由氩激光产生。通过插入屏障过滤器在500 nm以上记录发射。证据表明,该信号来源于RPE中的脂褐素,来自Delori及其同事的工作(84)他发现,脂褐素的光谱特征是典型的,信号源位于神经视网膜外部和脉络膜内部,这意味着其来源于RPE。使用SLO显示,在AMD早期,自身荧光的分布因患者而异。大约一半的自体荧光是均匀的,但其余部分呈弥散不规则或局部增强(85).德鲁森似乎无法解释这些差异,因为它们基本上不会自发荧光。研究表明,在双侧早期AMD中,自身荧光模式是对称的,这表明自身荧光特征反映了个体的疾病形式,可能由遗传或环境影响决定。在AMD导致单侧视力丧失的患者中,正常眼局部增强的自体荧光与另一只眼的GA相关,并预示着正常眼GA的后期发展。观察到在GA周围可以发现高水平的自体荧光,如果是这样的话,该区域在1年内就会萎缩,这进一步证实了这一结论(86).
RPE变化导致GA发展的潜在分子机制一直存在争议。有人认为,残体占据的细胞质体积可能会干扰细胞代谢(87).研究表明,脂褐素是一种自由基发生器,可能导致细胞损伤(88).此外,有证据表明,个别脂褐素成分具有毒性作用。A2-E在生物膜上具有类似表面活性剂的特性,一项研究表明,A2-E通过调节依赖ATP的溶酶体质子泵的影响来增加溶酶体内pH值。pH值的增加反过来会抑制溶酶体水解酶的活性(89).此外,A2-E已被证明能在体外引起溶酶体渗漏(90).溶酶体含量的释放可能导致RPE功能障碍和细胞死亡。另一项研究未能证实脂褐素存在时溶酶体pH值升高,可能是因为使用了较少的脂褐素,但它确实表明脂褐素降解减少了(91).因此,这两项研究都表明脂褐素降低了吞噬溶酶体酶的活性。
体内研究了RPE溶酶体降解减少的可能后果。通过玻璃体内注射溶酶体蛋白酶抑制剂E-64,对11周龄Sprague-Dawley大鼠的溶酶体降解产生干扰(92).单次注射导致RPE中吞噬小体样包涵体的短暂积聚。此外,每隔一天注射2或3次会导致这些内含物的进行性积累,这些与细胞内细胞器的变化有关,例如滑面内质网和RPE细胞构象的丢失。这伴随着光感受器外节的缩短和丢失,而之前没有畸形改变,光感受者丢失,脉络膜毛细血管窗孔减少,以及成纤维细胞和周细胞侵入Bruch膜。用于比较的玻璃体内注射载体未引起结构变化。
据认为,溶酶体蛋白酶抑制引起的RPE变化可能反映了脂质代谢减少、基底外侧血管内皮生长因子表达减少以及由此导致的脉络膜毛细血管窗孔丢失。外节的缩短和丢失被认为是由于椎间盘膜的形态发生受损引起的,而不是E-64对感光细胞的直接影响,因为椎间盘没有囊泡形成(92).外节缩短可能是由于RPE无法回收从吞噬体中解救出来的物质,导致可用脂质缺乏。这些发现表明,与之前考虑的相比,光感受器外节更新对吞噬体降解产物的可用性的依赖性更大,还表明获取血浆衍生材料不足以完全维持这一过程。RPE回收油脂的能力已得到很好的证明(93,94).在溶酶体蛋白酶抑制剂治疗的大鼠中观察到的变化在许多方面与RPE、感光细胞和脉络膜中与年龄相关的变化相似,尽管这种急性实验与终身代谢活动的后果以及大鼠和人类之间的物种差异存在重大差异。然而,实验证据和临床观察都说明了GA的潜在发病机制,并解释了如果后者不能循环吞噬体内容物,则其与局部增强的自体荧光的关联。
脂褐素存在的另一个潜在有趣的结果是,荧光团的光降解产物激活补体级联,这可能与Bruch膜增厚有关(95).因此,脂褐素可能通过多种机制导致与年龄相关的眼部变化和AMD。
对增加的自体荧光区域的视觉功能测量表明,暗视功能的丧失远大于明视功能,高达3.5 log单位(96).这种损失是由细胞丢失还是细胞功能障碍引起的还没有得到解决。
因此,有很好的证据表明,脂褐素增加对GA的形成很重要。鉴于维生素A对脂褐素的形成至关重要,膳食中补充维生素A可能是不可取的。通过限制维生素A对视网膜的可用性来减少脂褐素积累的治疗试验目前正在进行中。初步结果表明,这种方法在阿布卡4–/–老鼠(97).从理论上讲,限制光线也可能有帮助,因为视网膜的释放是由光线诱导的。增加溶酶体活性或降低吞噬溶酶体pH值的药物也可能有效,因为这可能抵消脂褐素对吞噬体内物质降解的影响。
AMD患者外视网膜的结构变化
相对于其他视网膜结构,关于AMD患者神经视网膜的物理变化的信息很少,其中感光细胞是AMD最重要的组成部分。早期组织学研究表明,早期AMD患者光感受器细胞的丢失是渐进性的,尽管人们认为这是RPE功能障碍的结果(98,99).临床研究支持了这一结论:2篇论文描述了使用一种称为眼相干断层扫描(OCT;refs)的专门成像技术对GA患者进行的研究结果。100,101).对萎缩边缘以外的眼底区域进行成像,以确定感光层的厚度,这些区域的视网膜在眼底镜下显示正常,除酒石外。在一些受试者中,从萎缩区缺乏感光细胞突然转变为正常厚度的外核层。然而,在大多数患者中,有证据表明,在萎缩边缘以外相当长的距离内,感光细胞大量丢失。因此,荧光素血管造影显示,在高自体荧光和慢脉络膜充盈区域记录的3个以上对数单位的功能丧失可能部分或全部由细胞丢失而非细胞功能障碍引起。因此,目前所有的证据都表明,即使在AMD早期,感光细胞的丢失也可能很严重,但不同个体之间的差异很大。
中的观察结果Cfh(立方英尺)–/–小鼠也可能与AMD患者的感光细胞丢失有关(75).与年龄匹配的动物相比,这些小鼠的视觉功能降低,尽管布鲁赫膜没有增厚。在这些小鼠中,光感受器外节畸形,外视网膜C3表达增加。目前尚不清楚C3与外节形态的相关性,以及这与交替补体途径失调对AMD发病机制的重要性这一信念之间的关系。根据这些观察结果,可以得出结论,CFH多态性与AMD高风险相关的后果可能不限于其对Bruch膜的影响。可能相关的是CFH公司在体外RPE中,似乎朝向顶端区域,这表明可能在视网膜外侧而不是脉络膜中表达(102).
虽然可以通过控制感光细胞外段A2-E的形成来减少脂褐素的积累,但通过改善RPE功能可以最好地保存感光细胞。限制维生素A的供应将达到这一目的。据推测,神经保护剂可能有益于青光眼的治疗(103).免疫系统与感光细胞健康的相关性尚不清楚,但有迹象表明,根据CFH的表达和视网膜外侧C3的存在,免疫系统可能很重要。当了解更多信息时,这可能是治疗的目标。
新生血管形成
有证据表明,脉络膜向内的血管生长是由生长因子失衡引起的(104).刺激生长的VEGF和抑制生长的色素上皮衍生因子(PEDF)受到了最大的关注。研究表明,RPE向脉络膜方向向外组成性表达VEGF,向神经感觉视网膜方向向内组成性表达PEDF(105).CNV患者的VEGF水平增加,PEDF水平降低(106,107).在光凝诱导的实验性CNV中,新的血管生长发生在应用后的头几天。几周内,RPE覆盖了病变的内表面,新血管关闭(108).在生长期,VEGF和PEDF的RPE表达分别增加和减少(109,110)在新血管复合体的自发分解过程中,情况发生了逆转。Yamagishi等人证明RPE负责控制血管反应的行为(111).在光凝后的不同时间间隔,将杀死RPE的碘酸钠注入玻璃体内。在应用激光时,没有出现新的血管形成。在激光应用4天后,这些船只继续在很长一段时间内增长,而不是像预期的那样关闭。因此得出结论,RPE产生的扩散因子对新血管生长的启动和随后的停堆都负有责任。
因此,这一概念已经形成,Bruch膜的正常非血管性质是由RPE抑制脉络膜血管向内生长引起的,并且这种变化是对年龄相关变化的响应。RPE对生长因子生产变化的刺激尚不清楚,但可能是由于缺乏血浆的代谢供应,这可能是由于物质通过加厚的Bruch膜扩散减少,或由于脉络膜毛细血管的变化导致代谢供应减少所致。
类似的机制可以解释视网膜血管瘤增生(RAP;参考文献。112).这似乎发生在早期AMD的环境中,涉及视网膜血管对视网膜外部的侵犯;目前尚不清楚它是否先于CNV或与CNV同时发生。正如生长因子可以阻止Bruch膜的血管化一样,它们也可以解释外层视网膜的无血管性。这包括生长因子第一次通过RPE细胞的基底外侧质膜域分泌,第二次通过顶端域分泌。因此,RAP可能是由顶端结构域而非基底侧质膜结构域引起的生长因子平衡变化引起的;或者,这两个域都可以发挥作用。
治疗试验表明,抗血管内皮生长因子药物对CNV患者的主要益处(113,114)尽管每月玻璃体内注射在人体工程学上很难实现。所有证据都表明,与大多数癌症的情况不同,新生血管的刺激是由单一扩散剂决定的。在早产儿视网膜病变和实验性视网膜动脉阻塞中的观察结果支持了VEGF是刺激眼新生血管的唯一或主要生长因子的结论,无论临床情况如何(115,116).
有趣的是,VEGF暴露于RPE的顶端区域会导致融合培养中的电阻降低,提示紧密连接完整性和某些极性的丧失(117).因此,抗血管内皮生长因子药物的治疗效果可能通过其对RPE和血管的作用来介导。
结论
在与年龄相关的疾病中,脉络膜、Bruch膜、RPE和外视网膜发生变化。基因和环境的影响都被认为具有疾病风险,并且可能是疾病的始作俑者。然而,对于这种组织复合体的哪种成分引发了这种疾病,仍存在疑问。改变的顺序可能因患者而异。如果是这种情况,确定任何个体中受影响最严重的组织都很重要,因为针对某一组织的治疗可能适合某些组织,但不适合其他组织。这需要准确的表型分析,这可能与自体荧光成像或OCT有关(118).然而,考虑到这些组织的代谢相互依赖性,一个组织中变化的调节必然会对邻近组织产生次要影响(图5).目前,有关于AMD终末期发病机制的概念,但对中间疾病机制知之甚少。从治疗的角度来看,在疾病发展的任何阶段进行干预都可能是有益的。与最近的情况形成鲜明对比的是,目前对各种致病过程的研究非常活跃。由于遗传学、细胞生物学和生物化学方面的成功,人们对疾病过程的认识不断加深。
图5。AMD的假定疾病途径,从过程的始发者到中间疾病过程再到最终事件。

始发事件被认为是由可能影响Bruch膜/脉络膜(BM/C)、RPE或感光细胞(PRC)的已知和未知遗传和环境因素产生的。组织的代谢相互依赖性使得任何一个组织的变化都不可避免地会影响其他组织,从而导致一系列中间疾病机制的复杂化,而这些机制目前尚不清楚。部分了解GA、CNV和PED的最终赛事。很可能在疾病发展的任何阶段进行有效的治疗干预都是有益的,并且针对一个组织的成功治疗将对其他组织产生有益的影响。
本综述并非详尽无遗,还考虑了非组织特异性的其他因素,如自由基损伤和线粒体功能障碍(119,120).有大量的间接证据支持每一种说法,但都没有得到证实。这一疾病的成功治疗迫在眉睫,它给西欧裔社区带来了巨大的健康负担,并有可能在其他社会中成为负担。鉴于本文所述的新信息和正在进行的调查,应在可预见的未来建立新型合理的患者管理技术。
致谢
作者感谢Dean Bok阅读了手稿并给出了有益的建议。
脚注
本文引文: 临床投资杂志。2010;120(9):3033–3041.doi:10.1172/JCI42437

