摘要
真核染色质被分为翻译后组蛋白修饰、组蛋白变体和DNA甲基化所分化的功能域1–6甲基化与植物和动物中转录起始的抑制有关,经常在转座因子中发现。正确的甲基化模式对真核生物的发育至关重要4,5甲基化诱导抑癌基因异常沉默是人类癌症的一个共同特征7与甲基化相反,组蛋白变体H2A。Z优先由Swr1-ATP酶复合体沉积在基因5′端附近,在那里它促进转录能力8–20DNA甲基化和H2A如何。Z影响转录在很大程度上仍然未知。我们在工厂中展示了这一点拟南芥,DNA甲基化区域在H2A中定量不足。Z.H2A除外。Z出现在活跃转录基因和甲基化转座子体内的DNA甲基化位点。MET1 DNA甲基转移酶突变,导致DNA甲基化的损失和获得4,5,在H2A中产生相反的变化。Z沉积,而沉积H2A的Swr1复合物的PIE1亚单位发生突变。Z轴17导致全基因组超甲基化。我们的发现表明,DNA甲基化可以影响染色质结构,并通过排除H2A影响基因沉默。Z、 和H2A。Z保护基因免受DNA甲基化。
调查H2A。Z沉积在植物染色质中,我们生成了H2A的高分辨率全基因组图。Z在拟南芥中的应用体内我们用于亲和纯化果蝇染色质的生物素化系统21.我们标记了拟南芥H2A。Z具有一种被特异性识别的肽大肠杆菌生物素连接酶BirA(生物素连合酶识别肽,BLRP),并创建共表达BLRP-H2A的转基因植物。Z和BirA。细胞定位显示BLRP-H2A。Z具有弥散的核分布,但被排除在异色色心之外(补充图1)与内源性H2A的模式相同。Z轴17.用微球菌核酸酶消化后,大部分为单核小体(补充图1),我们从根组织中纯化生物素化染色质,并将相关DNA与对照DNA在代表整个拟南芥基因组的贴片微阵列上进行联合杂交22为了确保我们的结果不受潜在标记伪影的影响,我们用内源性H2A抗体重复了实验。Z轴17我们还绘制了根中的DNA甲基化图(我们之前已经发布了来自气生组织的数据集22).
链霉亲和素下拉和免疫沉淀生成的图谱几乎相同(图1和补充图2).最显著的特征是与DNA甲基化强烈的定量反相关(Pearson’s第页= −0.81;补充表1-2).H2A的明显峰。基因5′端附近的Z也很明显(图1b).为了更好地可视化H2A。Z分布,我们将包括基因、假基因和转座元件在内的所有拟南芥注释序列在其5′端对齐,并将其从第1染色体顶部堆叠到第5染色体底部(图2a和补充图2).该路线的一个明显特征是高H2A的垂直条带。大致对应于转录开始后第一个核小体的Z。这种H2A模式。Z沉积与酵母和人类中的沉积一致10–15表明这是真核基因的一个普遍特征。还有五条明显的低H2A水平条纹。Z公司。这些对应于五个拟南芥着丝粒周围富含转座子、高度甲基化的异染色质。这种结合模式正好与DNA甲基化模式相反(图2b和补充图2).
图1。拟南芥H2A高分辨率图谱。Z和DNA甲基化。
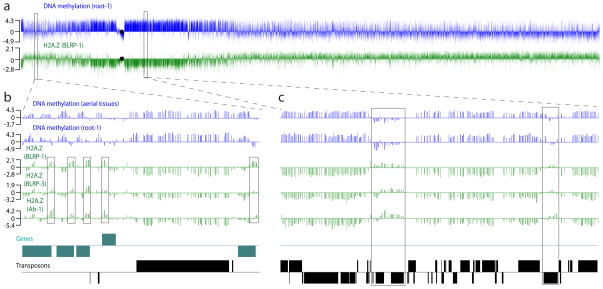
a、,H2A。拟南芥2号染色体的Z(绿色)和DNA甲基化(蓝色)图谱。每个竖线代表原木2测试样本信号除以输入控制信号的信号比(log2(测试/输入))。黑色圆圈表示着丝粒序列间隙的位置。b–c,常染色(位置547000–587000,b条)和一个异色(4407000–4463000,c(c))基因组区域。空中组织和根的DNA甲基化以蓝色显示;从两个独立的BLRP-H2A获得的HA2.Z剖面。Z转基因株系和内源性H2A的免疫沉淀。Z以绿色显示。线的上方和下方分别显示了顶部和底部链上的基因和转座子。H2A的5′峰。基因中的Z由中的方框强调b条H2A水平相对较高的非甲基化转座子。中的方框强调Zc(c).
图2。H2A。Z和DNA甲基化是相互排斥的。
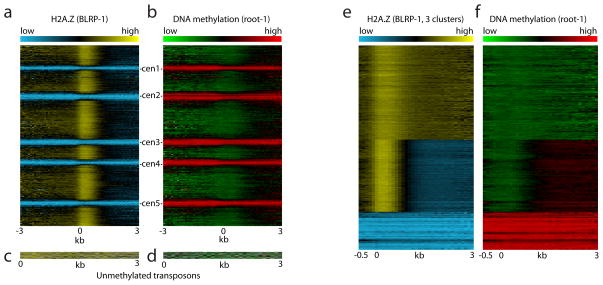
a–b,所有TAIR 7注释序列(31762)均在5′端对齐,并从1号染色体顶部堆积到5号染色体底部。BLRP-H2A。Z在中显示为热图一;根DNA甲基化显示为b条注意H2A之间的高度反相关。Z和甲基化。c–d,非甲基化转座元件(列于补充表3).BLRP-H2A。Z在中显示为热图c(c);根DNA甲基化显示为d、e、,所有TAIR 7注释序列均为k个-表示聚集(k个=3)基于BLRP-H2A。Z图案,并显示为热图。为了进行比较,相同序列的根DNA甲基化在(f).
甲基化在基因组中分布不均匀。转座子甲基化程度高且均匀,而一些基因甲基化时间短,而大多数基因是非甲基化的22–26这三组序列显示了H2A的相应三相分布。Z信号:低H2A。Z水平存在于转座子、甲基化基因的中间水平和非甲基化基因中(补充图3).一种可能性是H2A含量低。转座子中的Z是由内在序列偏好引起的,而不是DNA甲基化。为了测试这一点,我们检测了少量(49)未甲基化的拟南芥转座子(补充表3).很明显,所有这些转座子都具有高H2A。Z水平,表明H2A较低。Z合并本身不是转座子的特征(图1c和2c–d个).非甲基化转座子也缺乏任何可识别的H2A。Z峰值,表明这些是内源性基因的独特特征。无监督k个-表示基于H2A的拟南芥注释序列聚类。Z模式产生了三组与非甲基化基因、身体甲基化基因和转座子紧密对应的基因(图2e,补充图4和补充表4).同样是H2A。Z和DNA甲基化水平显示出显著的反相关(图2f).DNA甲基化和H2A。因此Z是相互排斥的染色质特征,我们的分析表明这种关系独立于序列上下文、转录或转录潜能(补充数据和补充图5-11).
到目前为止,我们的结果表明甲基化和H2A之间存在强烈的反相关。Z沉积,但我们无法区分哪个是因果关系。为了解决这个问题,我们利用了一个DNA甲基转移酶突变为零的株系MET1,金属1-64,27.中的突变MET1型导致整体DNA甲基化显著降低,但也会由其他甲基转移酶介导显著的超甲基化26我们推断,如果DNA甲基化影响H2A。Z沉积,DNA甲基化的变化应通过H2A的变化反映出来。Z分布。值得注意的是,因为满足1导致DNA甲基化的损失和获得,我们应该看到H2A的获得和损失。为了验证我们的假设,我们绘制了H2A图。Z、 以及DNA甲基化和转录金属1-6植物。
DNA甲基化的变化确实引起了H2A的变化。Z分布(图3和补充图12-13).为了可视化这些变化,我们减去了野生型(WT)H2A。来自的Z数据集满足1H2A。Z数据集,因此高值表示H2A增加。Z公司成立于满足1(补充图12).信息位点示例如所示图3a-c. The鱼类和野生动物管理局该基因通常具有5′甲基化并且缺少H2A。Z峰,失去启动子甲基化,获得5′H2A。Z英寸满足1(图3a).反转录转座子在5g13205在WT中严重甲基化,但失去甲基化并获得H2A。Z英寸满足1(图3b).基因在1g22000编码F-box蛋白,在满足1导致其5′H2A损失。Z峰值(图3c).
图3。H2A。Z合并变更金属1-6突变植物。
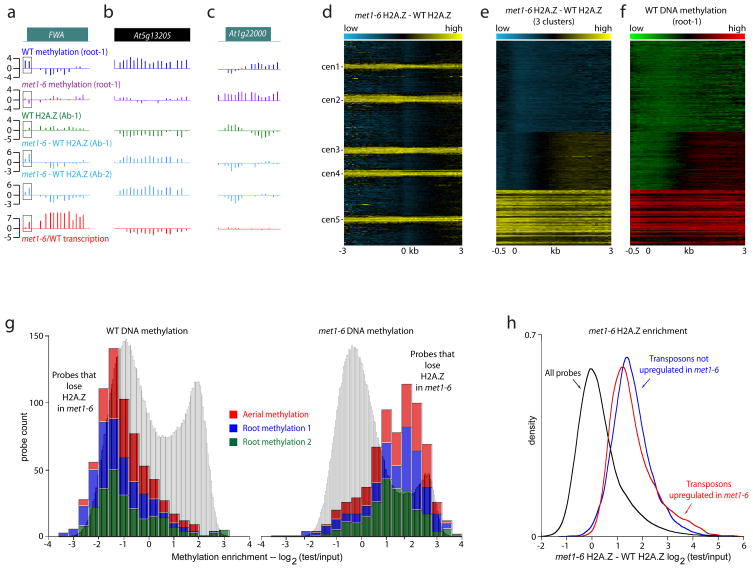
a–c,野生型(WT)根DNA甲基化(深蓝色),金属1-6根DNA甲基化(紫色),WT H2A。Z(抗体,绿色),WT H2A。从中减去Z轮廓金属1-6H2A。Z剖面(两组独立的配对实验,浅蓝色),以及金属1-6/WT转录(红色)鱼类和野生动物管理局在里面一,类copia转置元素在5g13205失去甲基化并获得H2A。Z英寸金属1-6在里面b条和F-box基因在1g22000它被高度甲基化并失去H2A。Z英寸金属1-6在里面c(c).的5′区域鱼类和野生动物管理局WT中的甲基化通过a.天,所有TAIR 7注释序列均在5′端对齐,并从1号染色体顶部到5号染色体底部堆叠。WT H2A。从中减去Z图案金属1-6H2A。Z图案显示为热图。之后的相同数据k个-意味着聚类(k个=3)如所示e(电子)为了进行比较,序列的根DNA甲基化排列如下e(电子)显示为中的热图f.g、,WT甲基化水平(左)和金属1-6探针的甲基化水平(右)表示H2A显著降低。Z英寸金属1-6(补充图12).三个独立的甲基化数据集的直方图是累积的。背景中的灰色直方图显示了所有探头的信号分布。小时,核密度图,用于跟踪数据集中显示的所有探针的频率分布d日(黑色痕迹),转座因子上调金属1-6(红色迹线)和转座因子在金属1-6(蓝色痕迹)。
全面了解H2A。Z动态输入金属1-6,我们对齐并排列了所有带注释的拟南芥序列,如图2a。在这个剖面上,同样明显的中心周围条纹也很明显(图3d和补充图13)–H2A。转座元件中的Z水平升高,它们失去了大部分甲基化,并在满足122,23数据的无偏分类产生了三个簇,分别大致包含非甲基化基因、甲基化基因和转座子(图3e,补充图13和补充表4,序列分类如下22).H2A的变化。Z与获得H2A的DNA甲基化序列密切相关。Z英寸满足1在WT中甲基化(图3f和补充图13).相反,H2A降低的位点。Z掺入在WT中未甲基化,但在金属1-6(图3g).总的来说,DNA甲基化的变化反映在H2A的变化上。Z的方式强烈证明甲基化抑制H2A。Z公司。
因为一些转座子和基因在满足1植物22,我们有机会测试H2A。Z掺入受甲基化负向影响或受转录正向影响。在基因内,WT和H2A中的DNA甲基化之间存在着强大的相关性。Z变化在金属1-6(平均皮尔逊第页= 0.51,补充表2),但转录与H2A之间没有相关性。Z变化(皮尔逊平均值第页= 0.05).鱼类和野生动物管理局,在满足1,降低了H2A水平。Z在基因体中,在WT中没有甲基化(图3a).类似地,在少数野生型中没有甲基化的转座子中,有两个(见4g10690和在5g35205)然而,在满足1(补充图14).两者的H2A也较少。Z英寸满足1而在野生型中,与其他转座子相反。
因为只有大约一半的转座元件在满足1,我们可以问这些元素是否优先获得H2A。Z、 如H2A所预期。Z掺入与转录活性相关。为了确保数据集的大小和甲基化不是问题,我们比较了代表活化转座子的12500个探针和代表沉默转座子并具有相同甲基化谱的12500种探针。我们发现这两个转座子类在H2A中的富集程度相同。Z轴(图3h和补充图15).因此,DNA甲基化的改变,而不是转录,导致H2A的重新分布。我们观察到的Z满足1.
我们的结果表明,DNA甲基化排除了H2A。一个有趣的问题是H2A是否存在。Z也可以排除甲基化。我们的一些数据表明,情况确实如此。H2A最显著的特点。Z掺入,即5′基因峰,与DNA甲基化无关(图2e–f和补充图4、6)然而甲基化被强烈排除在这个区域之外22,23同样,H2A越高。低转录基因体内的Z水平(补充数据和补充图6-7)这可能解释了令人费解的观察结果,即基因甲基化的几率随着转录的增加而增加(高达约70第个百分位数)22,23.
为了解决这个问题,我们绘制了植物中DNA甲基化的图谱项目1(Swr1的保守催化成分)破坏H2A的正常沉积。Z轴17。甲基化的总体模式图1-5植物仍与WT相似(补充表5),但DNA甲基化有适度但持续的增加(补充图16).可视化甲基化变化第1张我们从中减去匹配WT对照(F2同胞)的甲基化模式pie1并将结果数据显示为热图(图4a和补充图16).该分析揭示了基因体的全基因组超甲基化。使用ChIPOTle算法28,我们确定了1201个高甲基化区域(对应1172个基因)用于进一步分析(阈值p<10−7,补充表6).
图4。H2A。Z保护DNA甲基化。
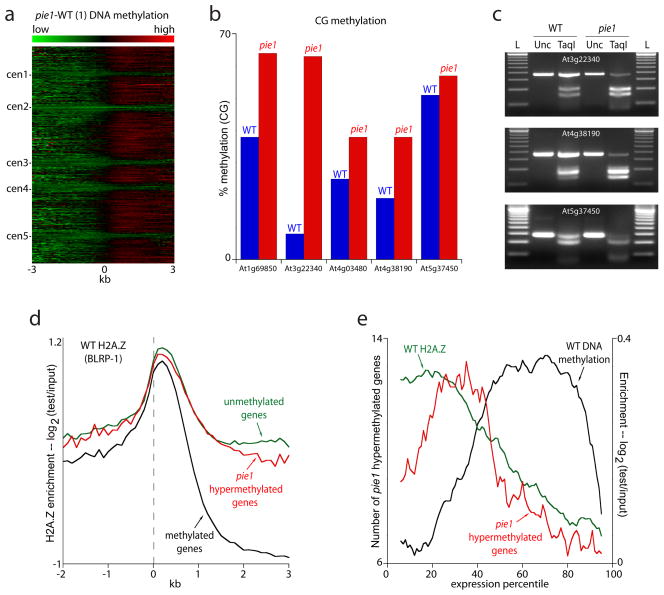
a、,所有TAIR 7注释序列均在5′端对齐,并从1号染色体顶部到5号染色体底部堆叠。WT甲基化模式从第1张甲基化模式显示为热图。b、,五个位点的亚硫酸氢盐测序结果。我们对每个基因型的12个克隆进行了测序,除了地址:1g69850(10个克隆第1张)和收件人4g38190(11个克隆第1张).c、,用TaqI消化来自亚硫酸氢转化基因组DNA的PCR产物,TaqI识别TCGA,只有在C未转化(因此甲基化)时才会切割。L=100 bp梯形图,Unc=未切割PCR产物,TaqI=用TaqI消化的PCR产物。请注意,在第1张与WT相比。日期:,所有基因在5′端对齐,每个100-bp间隔的平均分数从距离基因2 kb(负数)到基因3 kb(正数)绘制。使用5点滑动窗口对数据进行平滑处理。虚线表示对齐点。e、,基因根据转录水平分为百分位。红线追踪了在第1张在每个百分位内(左Y轴)。黑线表示DNA甲基化富集(所有基因),绿线表示H2A。非甲基化基因中的Z富集(右Y轴)。使用10点滑动窗口对数据进行平滑处理。右侧Y轴的刻度设置为从零开始,以便比较甲基化和H2A。Z.公司。
在植物中,任何胞嘧啶都可能发生DNA甲基化5大多数甲基化是在对称的CG位点中发现的,就像在动物中一样,是由MET1介导的,但在其他序列环境中也有大量的甲基化被其他甲基转移酶催化(因此在满足1)25,26。为了确定第1张突变在不同情况下影响DNA甲基化,我们使用亚硫酸氢盐测序分析了ChIPOTle评分为高甲基化的五个位点中单个胞嘧啶的甲基化:地址:1g69850(硝酸盐转运蛋白),地址:3G22340(类COPIA反转录转座子),收件人4g03480(含蛋白质的锚蛋白重复序列),收件人4g38190(纤维素合成酶)和地址:5g37450(一种蛋白激酶)。所有五个样本都显示CG甲基化适度但持续增加(图4b–c),确认微阵列分析。WT或WT中的任何一个位点几乎没有非CG甲基化第1张(未显示数据)。有趣的是,所有基因座在WT中都有一些甲基化,所以我们在第1张可能主要是由于正常轻度甲基化位点的甲基化增加,而不是从头开始先前未甲基化位点的甲基化。
鉴于第1张突变时,我们问过甲基化位点是否代表整个基因组。正如所料,第1张高甲基化基因含有高水平的H2A。WT中的Z(即通常在非甲基化基因中发现的Z;图4d).它们通常也富含低转录基因,在30岁左右含量最高第个转录百分位数(图4e).这种模式与正常的甲基化基因非常不同,甲基化基因在70岁左右最为普遍第个百分位数(图4e),也不同于非甲基化基因,后者富含低表达和高表达基因22.第1张然而,高甲基化基因确实与H2A的总体分布密切相关。Z轴(图4e).这些位点还包括49个富含H2A的转座子中的17个。Z和未甲基化的WT(补充表3、6),10倍的过度代表(p=10−4,费希尔精确测试)。因此,DNA甲基化的首选靶序列(基因体和转座子)在第1张与WT中这些序列中存在的低水平DNA甲基化一致(图4b–c).高水平的H2A。在这些位点上发现的Z明显保护了它们不发生完全的DNA甲基化,这可能解释了所观察到的基因转录和DNA甲基化之间的关系22.
几十年来,甲基化如何沉默基因一直是一个棘手的问题。一个流行的模型是,与甲基化DNA结合的蛋白质通过招募组蛋白去乙酰化酶而产生沉默6然而,对小鼠进行的仔细的基因破坏研究表明,这些蛋白质不太可能完全解释甲基化诱导的抑制29,30先前的研究已为H2A提供了强有力的证据。Z有助于提升推广人员的能力16–19因此,排除H2A。Z代表了DNA甲基化导致基因沉默的一种新机制。H2A。反过来,Z掺入可能保护基因启动子免受DNA甲基化的影响,从而促进基因活性并防止沉默。假设DNA甲基化和H2A。Z都是古老的染色质成分,它们的相互作用可能在调节真核基因表达中起着重要的普遍作用。
致谢
我们感谢Jorja Henikoff和Bao Nguyen对计算分析的帮助,Paul Talbert对细胞学的帮助,Terri Bryson和Andrew Morgan对技术支持,Mary Gehring和Brian Staskawicz对根培养方案的支持,Roger Deal和Richard Meagher对H2A的支持。Z抗体、用于进行微阵列杂交的FHCRC DNA阵列设施和用于转基因系的Martha Orozco。DC由NSF的产前研究金资助。DZ是白血病和淋巴瘤协会的研究员。
脚注
作者贡献D.Z.和S.H.构思了这项研究;D.Z和D.C进行了实验;D.Z.、T.B.、D.C.和S.H.分析了数据;D.Z.和S.H.写了这篇论文。
方法总结我们采用了我们在果蝇组织培养细胞中开发的生物素介导的亲和纯化系统21以便从拟南芥植物中纯化蛋白质。生物素化H2A。Z按所述进行了大量纯化21.内源性H2A。Z按所述进行免疫纯化17,但IP是在TNE中执行的。
我们的甲基化DNA IP协议(MeDIP)、微阵列设计和标记协议在22所有标记的样本均被送往NimbleGen Systems(威斯康星州麦迪逊)进行杂交,但第1张样本,在FHCRC DNA阵列设施进行杂交。对于亚硫酸氢盐测序,使用Qiagen EpiTect试剂盒对每个样品的2μg基因组DNA进行亚硫酸氢转化。
工具书类
-
1Malik HS,Henikoff S.核小体的系统发育学。自然结构生物。 2003;10:882–91.doi:10.1038/nsb996。[内政部] [公共医学] [谷歌学者]
-
2Bernstein E,Hake SB.核小体:微小的变异会带来很大的影响。生物化学细胞生物学。 2006;84:505–17.doi:10.1139/o06-085。[内政部] [公共医学] [谷歌学者]
-
三。Bhaumik SR,Smith E,Shilatifard A.发育和疾病发病过程中组蛋白的共价修饰。自然结构分子生物学。 2007;14:1008–16.doi:10.1038/nsmb1337。[内政部] [公共医学] [谷歌学者]
-
4Goll MG,Bestor TH。真核胞嘧啶甲基转移酶。生物化学年度收益。 2005;74:481–514.doi:10.1146/annurev.biochem.74.010904.153721。[内政部] [公共医学] [谷歌学者]
-
5Gehring M,Henikoff S.植物基因组中的DNA甲基化动力学。Biochim生物物理学报。2007年doi:10.1016/j.bbaexp.2007.01.009。[内政部] [公共医学] [谷歌学者]
-
6克洛泽RJ,伯德美联社。基因组DNA甲基化:标记及其介质。生物化学科学趋势。 2006;31:89–97.doi:10.1016/j.tibs.2005.12.008。[内政部] [公共医学] [谷歌学者]
-
7Feinberg AP、Ohlsson R、Henikoff S。人类癌症的表观遗传祖细胞起源。Nat Rev基因。 2006;7:21–33.doi:10.1038/nrg1748。[内政部] [公共医学] [谷歌学者]
-
8Mizuguchi G等。SWR1染色质重塑复合物催化的组蛋白H2AZ变体的ATP驱动交换。科学。 2004;303:343–8.doi:10.1126/science.1090701。[内政部] [公共医学] [谷歌学者]
-
9Guillemette B,Gaudreau L.重新整合H2A的对比功能。Z.生物化学细胞生物学。 2006;84:528–35.doi:10.1139/o06-077。[内政部] [公共医学] [谷歌学者]
-
10Guillemette B等,变异组蛋白H2A。Z全局定位于非活性酵母基因的启动子并调节核小体定位。《公共科学图书馆·生物》。 2005;3:e384。doi:10.1371/journal.pbio.0030384。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
11Li B,等。组蛋白变体H2AZ在非活性启动子上的优先占据影响局部组蛋白修饰和染色质重塑。美国国家科学院院刊2005;102:18385–90.doi:10.1073/pnas.0507975102。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
12Millar CB,Xu F,Zhang K,Grunstein M.H2AZ Lys 14的乙酰化与酵母的全基因组基因活性相关。基因开发2006;20:711–22.doi:10.1101/gad.1395506。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
13Raisner RM等人。组蛋白变体H2A。Z标记常染色质中活性和非活性基因的5′端。单元格。 2005;123:233–48.doi:10.1016/j.cell.2005.10.002。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
14Zhang H,Roberts DN,Cairns BR。Htz1的全基因组动力学,Htzl是一种组蛋白H2A变体,通过组蛋白丢失平衡受抑制/基础启动子以激活。单元格。 2005;123:219–31.doi:10.1016/j.cell.2005.08.036。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
15.Barski A等人。人类基因组中组蛋白甲基化的高分辨率分析。单元格。 2007;129:823–37.doi:10.1016/j.cell.2007.05.009。[内政部] [公共医学] [谷歌学者]
-
16Brickner DG等人H2A。Z介导的核外周基因定位赋予了先前转录状态的表观遗传记忆。《公共科学图书馆·生物》。 2007;5:e81。doi:10.1371/journal.pbio.0050081。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
17Deal RB、Topp CN、McKinney EC、Meagher RB。拟南芥开花的抑制需要组蛋白变体H2A激活开花位点C的表达。Z.植物细胞。 2007;19:74–83.doi:10.1105/tpc.106.048447。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
18Meneghini MD、Wu M、Madhani HD。保守组蛋白变体H2A。Z保护常染色质免受沉默异染色质的异位扩散。单元格。 2003;112:725–36.doi:10.1016/s0092-8674(03)00123-5。[内政部] [公共医学] [谷歌学者]
-
19Updike DL,Mango SE。HTZ-1/H2A对前肠发育的时间调节。Z和PHA-4/FoxA。公共科学图书馆-遗传学。 2006;2:e161。doi:10.1371/journal.pgen.0020161。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
20Venkatasubrahmanyam S,Hwang WW,Meneghini MD,Tong AH,Madhani HD。与局部抗沉默相反,全基因组抗沉默是由常染色因子Set1和H2A冗余介导的。美国科学院Z.Proc Natl Acad Sci US A.2007;104:16609–14.doi:10.1073/pnas.0700914104。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
21Mito Y,Henikoff JG,Henikoff S.组蛋白H3.3替换模式的基因组尺度分析。自然遗传学。 2005;37:1090–7.doi:10.1038/ng1637。[内政部] [公共医学] [谷歌学者]
-
22Zilberman D、Gehring M、Tran RK、Ballinger T、Henikoff S.拟南芥DNA甲基化的全基因组分析揭示了甲基化和转录之间的相互依赖性。自然遗传学。 2007;39:61–9.doi:10.1038/ng1929。[内政部] [公共医学] [谷歌学者]
-
23张欣,等。拟南芥DNA甲基化的全基因组高分辨率定位和功能分析。单元格。 2006;126:1189–201.doi:10.1016/j.cell.2006.08.003。[内政部] [公共医学] [谷歌学者]
-
24Vaughn MW等人,《拟南芥的表观自然变异》。《公共科学图书馆·生物》。 2007;5:e174。doi:10.1371/journal.pbio.0050174。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
25Cokus SJ等。拟南芥基因组的Shotgun亚硫酸氢盐测序揭示了DNA甲基化模式。自然。 2008;452:215–9.doi:10.1038/nature06745。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
26李斯特R等,《拟南芥表观基因组高度整合的单基分辨率图谱》。单元格。2008年doi:10.1016/j.cell.2008.03.029。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
27肖伟,等。MEA Polycomb基因的印迹是由MET1甲基转移酶和DME糖苷酶之间的拮抗作用控制的。开发单元。 2003;5:891–901.doi:10.1016/s1534-5807(03)00361-7。[内政部] [公共医学] [谷歌学者]
-
28Buck MJ、Nobel AB、Lieb JD。ChIPOTle:用于分析ChIP芯片数据的用户友好工具。基因组生物学。 2005;6:R97。doi:10.1186/gb-2005-6-11-r97。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
29Guy J、Hendrich B、Holmes M、Martin JE、Bird A。一种小鼠Mecp2-null突变会引起类似Rett综合征的神经症状。自然遗传学。 2001;27:322–6.doi:10.1038/85899。[内政部] [公共医学] [谷歌学者]
-
30Hendrich B、Guy J、Ramsahoye B、Wilson VA、Bird A。密切相关的蛋白MBD2和MBD3在小鼠发育中起着独特但相互作用的作用。基因开发2001;15:710–23.doi:10.1101/gad.194101。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。

