摘要
细胞衰老被理论化为对抗由致癌途径激活引发的肿瘤转化在体外1–三,但衰老的相关性体内尚未建立。PTEN和p53抑癌基因是包括前列腺癌在内的人类癌症中最常见的失活或突变基因4,5虽然它们在功能上是不同的,但已经提出了相互合作的观点,因为PTEN被认为调节p53的稳定性,而p53被认为增强PTEN公司转录6–10这里我们显示了Trp53基因在小鼠中,前列腺不能产生肿瘤表型,而完全铂族前列腺失活在长潜伏期后引发非致死性浸润性前列腺癌。引人注目的是,联合灭活铂族和Trp53基因早在青春期后2周就会引发侵袭性前列腺癌,在7个月大的时候总是致命的。重要的是,急性铂族失活通过p53依赖性细胞衰老途径诱导生长停滞在体外和体内,可通过综合损失Trp53基因此外,我们在早期人类前列腺癌标本中检测到细胞衰老的证据。我们的结果证明了细胞衰老在限制肿瘤发生中的相关性体内并支持一种合作肿瘤抑制模型,其中p53是一种重要的失效保护蛋白铂族-缺乏肿瘤。
“细胞衰老”描述了原代培养细胞中细胞周期停滞的一种永久形式,可由DNA损伤或激活的致癌基因触发1–三虽然它与调节抗肿瘤治疗反应有关11,目前还没有证据表明衰老与肿瘤发生有关。
多达70%的原发性前列腺肿瘤会失去一个PTEN公司等位基因并保留另一个拷贝12–15类似地,p53几乎只在晚期前列腺癌中完全缺失或突变16,17.因为完全失去铂族在小鼠中似乎对侵袭性前列腺肿瘤的发展至关重要18,19,为什么人类侵袭性前列腺癌也不会选择PTEN公司-损失令人费解。同样,p53在晚期前列腺癌中丢失而不是在表现时丢失的观察结果令人惊讶,因为p53的丢失有利于许多组织的肿瘤发生。一个涉及PTEN和p53相互依赖的网络已经出现,可以部分地调和这个悖论。具体来说,PTEN可以保护p53免受Mdm2介导的降解,而p53可以增强PTEN的转录6–10任何一种基因的失活都会导致另一种基因蛋白质水平降低。因此PTEN公司或者p53可以模拟一个基因中的两个基因点击。为了在一个定义明确的肿瘤进展模型中测试这些观察结果的相关性,我们制作了小鼠,其中铂族和/或Trp53基因前列腺中的基因被特异性删除。这一分析导致了意外的观察结果,这些观察结果与上述假设相矛盾,并表明了细胞衰老对肿瘤抑制的重要性体内.
对于前列腺特异性失活,我们使用Cre/液氧磷技术20和普罗巴斯因-Cre4(Pb-Cre4号机组)转基因小鼠表达Cre公司青春期后(7周龄)前列腺上皮21。我们获得了铂族液氧磷/液氧磷;Pb-Cre4号机组和Trp53基因液氧磷/液氧磷;Pb-Cre4号机组小鼠,以下简称铂族pc−/−和Trp53基因pc−/−(补充图S1).不出所料,在Pb-Cre4号机组,重组铂族和Trp53基因局限于三个前列腺叶,即前前列腺(AP)、腹前列腺(VP)和背外侧前列腺(DLP),精囊中发生轻微重组(补充图S2a).
研究早期效应铂族和/或Trp53基因前列腺失活,9周龄时处死小鼠并进行组织病理学检查进行分析。野生型(WT)小鼠前列腺组织学正常,而年龄匹配的铂族pc−/−同寝的人始终如一所有三个前列腺均显示为高级别前列腺上皮内瘤变(HG-PIN)肺叶(大约50-60%的前列腺受到影响:n个= 10;图1a和补充图S2b),显示肿瘤发生的早期铂族切除。在Trp53基因pc−/−小鼠,无病理变化(增生或PIN)见于任何前列腺叶,其前列腺与年龄匹配的WT小鼠无明显区别(n个= 10;图1b和补充图S2b).最多18个月(补充图S2c未显示),雄激素受体(腺上皮标记物)或p63(基底细胞标记物)的免疫染色在形态上没有差异,也没有检测到差异(补充图S2d).
图1。损失Trp53基因不会引发前列腺肿瘤,但会导致铂族-致命的缺陷癌。
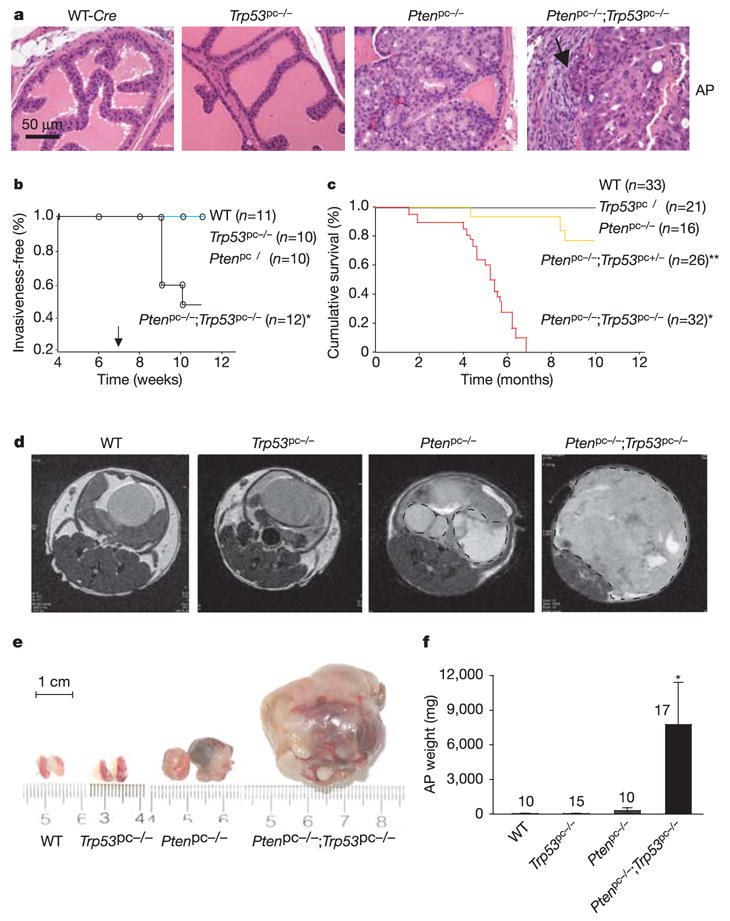
一,WT中前列腺前部(AP)的组织病理学分析(苏木精/伊红染色),铂族和Trp53基因9周龄时的单突变体和双突变体显示WT和Trp53基因pc−/−小鼠,但PIN损伤铂族pc−/−小鼠与入侵铂族pc−/−;Trp53基因pc−/−老鼠。b条前列腺癌的无病生存曲线(Kaplan–Meier图)。腺癌仅见于铂族pc−/−;Trp53基因pc−/−队列(P(P)< 0.05).箭头表示青春期。c(c),累积生存分析。寿命显著缩短(P(P)<0.0001)与铂族pc−/−队列是为铂族pc−/−;Trp53基因pc−/−队列(星号)和铂族pc−/−;Trp53基因个人电脑+/-队列(双星)。d日–(f)23-31周时AP肿瘤的MRI(虚线圆圈)(d)和它们的实际尺寸(e)和重量(f)活检后。中的误差线(f)指示杆上方所示小鼠数量的AP重量标准差。
相反,在联合灭活铂族和Trp53基因,我们在100%的小鼠的所有三个肺叶中都发现了HG-PIN,并且50%的患者患有侵袭性前列腺癌铂族pc−/−;Trp53基因pc−/−10周龄小鼠(n个= 12) (图1a、b和补充图S2b).11周时,浸润性腺癌仅限于铂族pc−/−;Trp53基因pc−/−突变体(图1b)然而铂族pc−/−动物在潜伏期4-6个月后出现浸润性前列腺癌。因此,与损失相比铂族,损失Trp53基因不会引发前列腺肿瘤,但会加速因失去铂族.
我们在10个月内对128只小鼠进行了生存分析两周磁共振成像(MRI)分析19。没有铂族pc−/−突变体死于前列腺癌(图1c),这与我们之前的研究一致19相比之下Trp53基因在一个铂族-无背景显著降低生存率,呈剂量依赖性(图1c).值得注意的是,所有铂族pc−/−;Trp53基因pc−/−小鼠在7个月大时死亡(平均存活5±0.5个月),可能是由于膀胱梗阻和肾功能衰竭。在这些小鼠或铂族pc−/−2.5年随访的小鼠(参考。19、Z.C和P.P.P.,未发表的观察结果)。到6个月时,MRI显示铂族pc−/−前列腺,但不在Trp53基因pc−/−或野生型前列腺(图1d).双阴性小鼠的平均肿瘤重量比铂族 pc−/−老鼠(图1e、f)肿瘤主要表现为低分化组织学(补充图S2c).肿瘤发生于铂族pc−/−;Trp53基因pc−/−小鼠包括整个泌尿生殖道和骨盆。相反,肿瘤铂族pc−/−小鼠的泌尿生殖器官边界清晰可辨。因此Trp53基因导致大量肿瘤生长和致命的前列腺癌,但只能与铂族缺乏。
我们研究了Trp53基因和损失铂族采用原代小鼠胚胎成纤维细胞(MEFs)。学习急性灭活铂族和Trp53基因我们用绿色荧光偶联的表达Cre-PURO-IRES的逆转录病毒感染MEF蛋白质(GFP)(图2a和补充图S3a).病毒介导的Cre表达导致loxP等位基因和增加Akt激活(补充的图S3a).铂族杂合的(铂族Δ /+)MEF的扩散速度比铂族重量(铂族+/+)MEF,但扩散铂族-空(铂族Δ/Δ)MEF与WT相似(图2a).铂族-空MEF显示了衰老细胞的独特形态(扁平大细胞;未显示)和衰老相关β-半乳糖苷酶(SA-β-Gal)阳性,这是衰老细胞22(图。2a个和补充图。S5a系列).确定衰老是否与选择过程或逆转录病毒整合,我们通过腺病毒核心(Ad-Cre)感染。我们发现失去一个铂族与野生型细胞相比,等位基因导致了细胞的加速生长二者都铂族等位基因导致低增殖和高β-Gal染色(补充图S3b、c和S5d).为了确保这些发现与Cre活动无关,我们尝试铂族通过RNA干扰(RNAi)击倒。杂合子MEF中Pten水平降低50%确实导致增殖和衰老诱导降低,不仅证实了Cre对衰老的依赖性,而且还表明细胞急性丧失铂族低于杂合阈值可以引起细胞衰老反应(图2b、和补充图S3d和5亿美元).
图2。急性损失铂族触发原代小鼠胚胎成纤维细胞(MEFs)p53依赖性衰老途径。
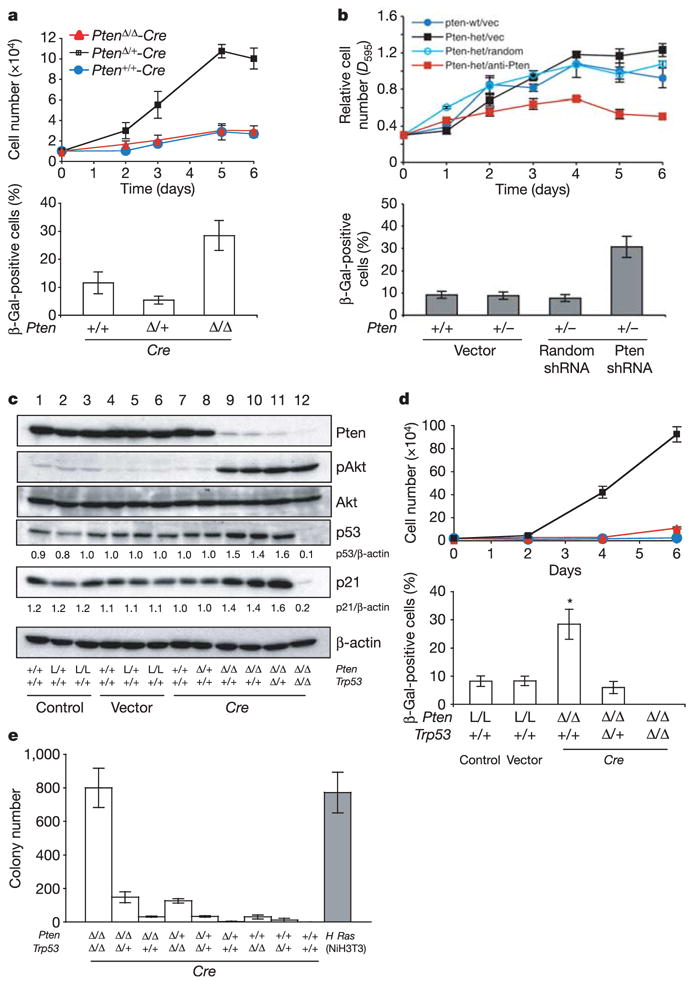
一Top:原发性MEF的生长曲线,感染逆转录病毒Cre(有选择)并随访6天。底部:来自a.乙,Top:感染Pten shRNA(有选择)的原代MEF的生长曲线,并在6天内进行跟踪。底部:来自b、cMEF裂解物的Western blots。数字表示与β-肌动蛋白相关的密度测定蛋白质水平。d日、生长曲线(顶部)和衰老染色(底部)铂族Δ /Δ;Trp53基因Δ /Δ双零MEF(黑色方块),铂族Δ /Δ;Trp53基因Δ /+MEF(红色三角形)和铂族Δ /ΔMEF(蓝色圆圈)。误差线表示s.d。升/升表示铂族液氧磷/液氧磷.e(电子),所有组合的转化分析铂族和Trp53基因灭活。H-Ras感染的NIH 3T3细胞作为阳性对照(灰色条)。误差条表示代表性实验的标准偏差,一式三份。
接下来我们评估了p53衰老途径的状态。急性损失铂族导致Akt激活并持续诱导p53水平(图2c)而不是p53丝氨酸15的磷酸化(补充图S3f).p21和衰老标志物纤溶酶原激活物抑制剂-1(PAI-1)的上调在中观察到铂族-MEF不足,但不在铂族/Trp53基因双零MEF(图2c和补充图S3g).此外,损失Trp53基因导致了铂族-背景为空(图2d和补充图S3e).此外,SA-β-Gal活性铂族Δ/ΔMEFs以p53剂量依赖的方式急剧下降,在双阴性MEFs中无法检测到。这些发现证实,逃避衰老对于铂族Δ /ΔMEF实现充分增殖潜力(图2d).TdT-介导的dUTP缺口末端标记(TUNEL)染色显示,两组之间的细胞凋亡无差异铂族-null和铂族/Trp53基因双空MEF(2天和4天,未显示)。最后,我们测试了p53是否对抗MEF的细胞转化23.完全灭活铂族和Trp53基因导致MEF转化,而转化电位在铂族或Trp53基因复制相关方式(图2e).这些发现表明,p53介导的衰老反应在细胞失去铂族.
接下来,我们研究了p53的稳定性是否因丢失铂族如前所述9.英寸事实上,我们观察到p53蛋白的半衰期延长而不是缩短铂族-与WT MEF相比为空(图3a).这可以用以下事实来解释:铂族导致p19的积累增加阿尔夫(图3b)它通过Mdm2抑制诱导细胞衰老和p53稳定1,2第16页INK4a蛋白质水平也增加了(图3b).特别是我们发现p21的半衰期延长了(图3a)表明p53诱导依赖(转录后)机制也可能影响p21的积累。然而,p53对p21的诱导至关重要,因为p21在第53页/铂族双空单元格(图2c).此外,如前所述24,一种激活形式的Akt(肉豆蔻酰化Akt)在原代MEF中引发生长停滞和细胞衰老,支持了以下观点:衰老是由铂族至少部分是由于Akt激活(图3c和补充图S5c).
图3。急性损失铂族结果在原发性MEF中ARF上调和p53/p21稳定。
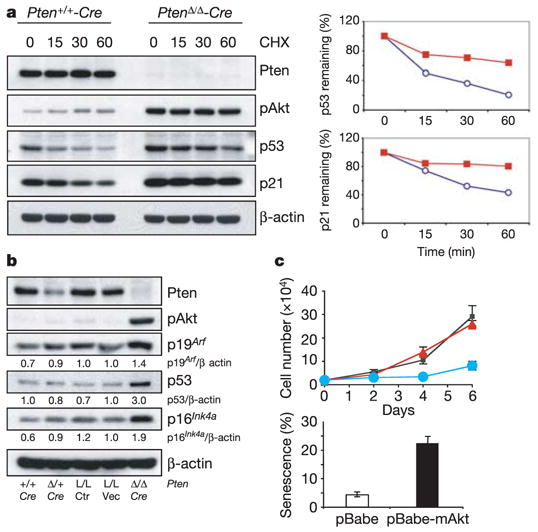
一左:环己酰亚胺(CHX)和western印迹在指定时间(分钟)对蛋白质合成的抑制表明,在MEF中Pten急性丢失时,p53和p21蛋白的半衰期延长。右图:从蛋白质印迹定量p53和p21半衰期,归一化为β-肌动蛋白。蓝色圆圈,铂族+/+-Cre公司;红色方块,铂族Δ /Δ-Cre公司.b条,Western blotting显示p19上调阿尔夫急性期后蛋白质铂族MEF损失。铂族+/+-Cre公司,铂族Δ /+-Cre,铂族液氧磷/液氧磷(L/L,Ctrl)和铂族液氧磷/液氧磷-MSCV载体(Pten升/升-vec)作为对照。c(c)MEF中肉豆蔻酰化Akt(mAkt)的表达诱导生长停滞(顶部)和细胞衰老(底部)。黑色方块,控制;红色三角形,pBabe;蓝色圆圈,pBabe-mAkt。误差条表示代表性实验的标准偏差,一式三份。
确定这些观察结果是否相关体内到我们对前列腺癌进行了免疫组化分析(IHC)。Pten染色那个铂族被有效且明确地删除,并且不受Trp53基因状态(补充图S4a).Akt活化和膜募集仅与以下损失相关铂族在里面铂族pc−/−和铂族pc−/−;Trp53基因pc−/−小鼠,而非p53状态(补充图S4a).同时,冷冻前列腺切片β-Gal活性染色。鉴于铂族-含无效前列腺大量的衰老细胞(“蓝色腺体”),WT前列腺很少(图4a和补充图S6b).量化显示与WT相比,11周时上皮细胞衰老(图。4b个),但双失活前列腺中的衰老细胞明显减少(图4a、b和补充图S6b).此外,没有细胞衰老杂合子灭活后检测铂族在里面铂族个人电脑+/-单个或复合突变体(补充图S6a),这是与我们的MEF结果一致(补充图S5a、b、d)并完全同意以下事实:Trp53基因没有加速PIN表型铂族个人电脑+/-突变体(未显示)。不同突变体前列腺的细胞凋亡保持可比性(图4c)而上皮细胞的衰老伴随着细胞增殖的反向变化(图4d);这已通过双重标记得到证实铂族-Ki-67和β-Gal突变腺体和双突变腺体(补充图S4b).
图4。p53依赖性细胞衰老途径限制肿瘤发生铂族-前列腺缺乏。
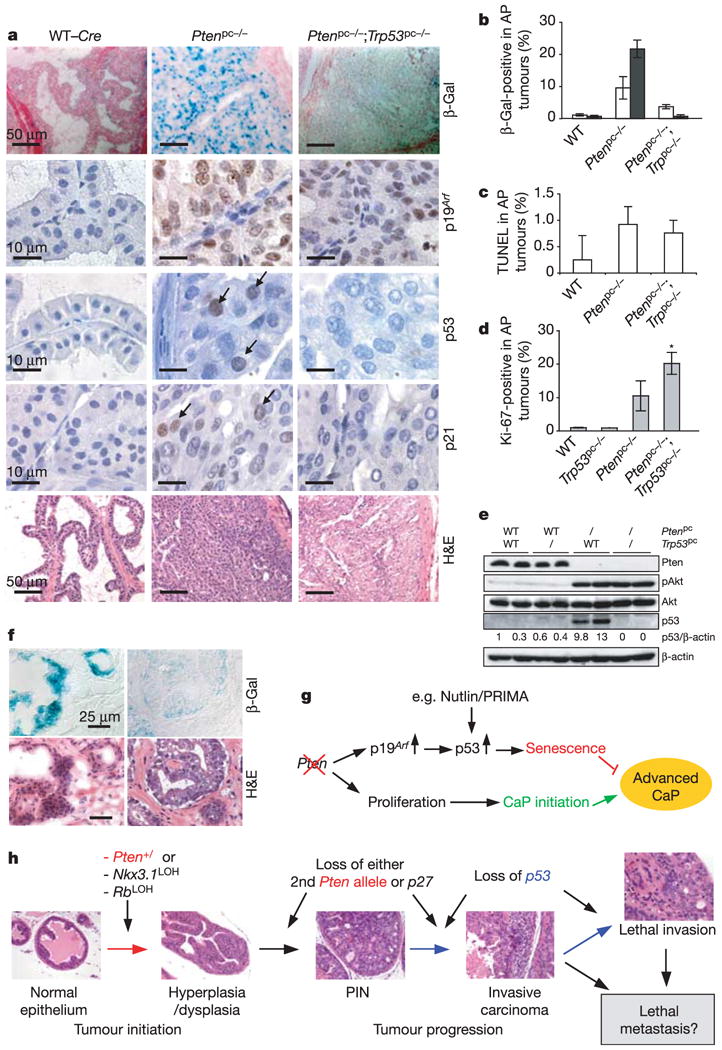
一11周龄前列腺的衰老和组织病理学分析,染色如图所示。H&E、苏木精/曙红。比例尺,50μm(β-Gal和苏木精/伊红染色)和10μm(p53、p19阿尔夫和第21页)。b条第8周(开放条)和第11周(填充条)AP切片上β-Gal染色的定量。对每种基因型的三只小鼠代表性切片进行计数。c(c),11周时AP细胞凋亡的TUNEL定量分析(每个基因型两只以上的小鼠)。d日,对11周龄AP进行Ki-67染色定量,如b。中的误差线b条–d日代表s.d.进行代表性实验,一式三份。星号表示铂族pc−/−和铂族pc−/−;Trp53基因pc−/−双突变体(P(P)< 0.05).e(电子)Western blot分析11周时每个基因型的AP组织显示Akt激活和p53诱导铂族pc−/−老鼠。(f)人前列腺癌根治术标本冷冻切片的衰老(β-Gal)和组织病理学(苏木精/伊红染色)分析显示,增生(癌前)前列腺(左侧)的β-Gal染色强,另一患者的肿瘤腺体(右侧)的β-Gal染色弱。比例尺,25μm。克,一个模型铂族-肿瘤发生缺陷、p53协同性及其治疗意义。铂族-用恢复突变p53活性的药物(例如PRIMA)治疗缺陷肿瘤可能会受益28)或通过灭活MDM2(例如Nutlin)来稳定WT p5329).小时,前列腺肿瘤发生、发展和进展的模型由以下因素协同贡献铂族,Trp53基因和其他基因。Trp53缺失通过衰老逃避机制加速癌症进展铂族-缺乏肿瘤。
接下来我们研究了p53是否上调铂族灭活体内事实上,我们发现p53在前列腺上皮细胞核中的积累增加铂族pc−/−小鼠,但不来自WT(或铂族pc−/−;Trp53基因pc−/−老鼠(图4a).裂解液来自铂族pc−/−免疫印迹法显示前列腺p53的诱导倍数超过10倍(图4e).重要的是,第19页阿尔夫p21蛋白水平在铂族pc−/−老鼠(图4a),确认体内激活p19阿尔夫→p53→p21衰老途径。
为了解决这些发现与人类癌症的相关性,我们对衰老进行染色在早期人类前列腺癌标本中(n个= 12;补充表S1).前列腺区域特异性观察到强β-Gal染色某些腺体增生/PIN(12个样本中的3个,放大倍数×100),但从未出现弗兰克肿瘤区域。高倍镜(×400)下,在另外四个样本中观察到弱染色,主要在前列腺增生/PIN区域,很少在癌区域(图4f;补充表S1).
前列腺癌是男性癌症相关死亡的第二大原因(仅次于肺癌),在美国每六名男性中就有一人罹患前列腺癌25,26涉及多种肿瘤抑制基因的改变27我们的数据通过证明Pten和p53在前列腺中的缺失,为深入了解Pten与p53之间的分子关系提供了依据铂族诱导p53功能(图4g).由于还不清楚p53功能的丧失是否与前列腺癌的发生或进展有关,我们的数据坚定地将p53功能丧失与前列腺癌发病或进展相关Trp53基因在前列腺癌进展中起关键作用(图4h).我们的结果表明铂族不会导致p53水平和功能降低,但会增加。相反,损失Trp53基因对Pten表达式没有影响在体外在前列腺上皮中,同时伴有铂族观察到前列腺癌的致死加速。因此,关于Pten–p53网络,我们的数据不支持肿瘤发生的“二合一”命中模型,但由于两个基因都需要消融以实现最大的疾病进展,因此他们建议对这些主要肿瘤抑制基因的遗传交互作用采用“一对一”命中模式。我们的发现具有治疗意义,因为它们表明PTEN公司-缺乏前列腺癌可能受益于增强p53激活的药物,有利于细胞衰老计划28,29(图4g).急性纯合子(非杂合子)缺失铂族触发p53依赖性细胞衰老程序体内提供了一个似是而非的解释,解释了为什么人类前列腺癌在呈现时没有选择完全丧失PTEN公司从而突出了PTEN单倍体不足与前列腺癌发病的相关性。此外,单独的p53完全失活在前列腺中没有表型后果的发现可以解释为什么第53页优先选择用于晚期前列腺癌,除此之外,它还可以通过以下途径使肿瘤达到最大增殖PTEN公司杂合性缺失。
方法
铂族和Trp53基因突变小鼠
铂族液氧磷/液氧磷小鼠的产生如前所述19、和Trp53基因液氧磷/液氧磷老鼠是用类似的策略生成(补充图S1;详细信息可从作者处获得)。女性铂族液氧磷/液氧磷;Trp53基因液氧磷/液氧磷小鼠与雄性杂交PB-核心4转基因小鼠21前列腺特异性缺失铂族和Trp53基因为了进行基因分型,用以下引物对尾部DNA进行聚合酶链反应分析。对于铂族液氧磷/loxP使用引物1(5′-AAAAAGTTCCCCTGCTGATGATTGT-3′)和引物2(5′-TGTTTTGACCATTAAAGTAGGCTGTGGTG-3′)。为了检测缺失的等位基因,使用了Lpten3引物(5′-TTCTCTTAGCA CTGTTTCACAGGC-3′)和引物1。对于Trp53基因液氧磷/液氧磷,引物1(5′-GAGACGCTGCCGGTTCCCTCC-3′)和引物2(5′-GCAAGA GGTGACTTGAAGCTC-3′)被使用。
构件
pMSCV-Cre-PIG通过亚克隆构建Cre公司pMSCV-PURO-IRES-GFP(MSCV-PIG)载体(S.W.Lowe赠送)。核定位序列融合Cre重组酶(NLS-Cre)从pTZ-CreN载体(来自L.Nitschke的礼物)中切除。
细胞增殖、转化、衰老和凋亡检测
如前所述,从不同基因型的单个胚胎中制备初级MEF30感染表达Cre-PURO-IRES-GFP、肉豆蔻酰化Akt(mAkt)或相应对照病毒的逆转录病毒。制备逆转录病毒颗粒,2×106将Phoenix细胞置于10 cm培养皿中,然后用Lipofectamine 2000(Invitrogen)转染pMSCV-Cre-PURO-IRES-GFP或空载体。对于感染,第2代的MEF以(3-4)×10的密度进行电镀5每10厘米培养皿培养一个细胞,转染48小时后被凤凰细胞中的病毒感染。在3μg ml存在下选择后−1利用嘌呤霉素(Sigma)、第6代MEF进行生长曲线、western blotting和衰老分析。为了测定衰老,在104用衰老检测试剂盒(Calbiochem)检测SA-β-Gal活性,并进行定量(每个样品200多个细胞)。对于前列腺组织,6μm厚的冰冻切片按上述方法进行β-Gal染色。对于转化分析,选择MEF(3×104)在传代6时,将其悬浮在含有0.3%琼脂的培养基中,每孔六孔板上的固化琼脂为0.6%,21天后评估菌落数。为了进行细胞凋亡分析,脱蜡和再水化石蜡切片用就地细胞死亡检测试剂盒(罗氏)。TUNEL阳性染色鉴定凋亡细胞。每个部分从五个不同的区域总共计数500个细胞,每个基因型至少重复两次分析。
短发夹RNA(shRNA)
利用寡聚引擎RNAi设计工具设计了Pten-directed shRNA(5′-GATCCACAGACCA TAACCCACAGTTCAAGAGACTGGTGGTGTGTGT ATGGTCTTA-3′);将产生的寡核苷酸亚克隆到pSUPER-puro载体(寡核苷酸引擎)中,并转染到Phoenix包装细胞中。以随机寡核苷酸(5′-GATCCCCAAGAGAGACGAGAAGAATTCAGATTCGTC TCCTTTTTA-3′)为对照。主要Pten第2代的WT或杂合子MEF被感染,选择(2μg ml中3天−1嘌呤霉素)并在2.5×10的条件下进行电镀412孔培养皿中每孔的细胞数为两个,用于通过光谱测量结晶紫吸收测定生长曲线。在生长曲线的第4天用相同和平行处理的细胞测量β-Gal活性。
蛋白质印迹和免疫组织化学
MEF裂解物用RIPA缓冲液(1×PBS、1%非奈特P40、0.5%脱氧胆酸钠、0.1%十二烷基硫酸钠和蛋白酶抑制剂鸡尾酒(Roche))制备。以下抗体用于免疫印迹:小鼠单克隆抗Pten(6H2.1;级联生物科学)、兔多克隆抗p19阿尔夫(Ab-1;钙生物化学)、兔多克隆抗p53(CM5;Novocastra)、兔抗Akt多克隆抗体和Akt的抗磷丝氨酸473(细胞信号)、兔多克隆抗p21(C-19;Santa Cruz)、兔耐p16多克隆抗体(M-156;Santa Cruz),兔多克隆抗体PAI-1(H-135;Santa Cruz)和小鼠单克隆抗β-肌动蛋白(AC-74;Sigma)。测定p53/p21蛋白半衰期,MEFs(铂族+/+-Cre公司和铂族Δ /Δ-Cre公司,在80%的合流和相同的传代数下)用30μg ml处理−1环己酰亚胺(Sigma),并在指定的时间采集用于蛋白质印迹分析。为了进行前列腺分析,通过在RIPA缓冲液中研磨前列腺组织,以1 mg/5μl的比例制备蛋白质提取物。经过短暂的超声和离心后,收集上清液进行蛋白质印迹。使用的抗体是兔多克隆抗PTEN(9552;细胞信号转导)。针对p53、pAkt和Akt的抗体如上所述。对于免疫组织化学(IHC),按照标准程序将组织固定在10%福尔马林中并嵌入石蜡中。对切片进行磷酸化Akt(Ser 473)抗体(细胞信号)、PTEN(Ab-2;NeoMarkers)、Ki-67(Novocastra)、p19的染色阿尔夫(ab80-50;Abcam)、p21(F-5;圣克鲁斯)、p53(FL-393;圣克鲁兹)、雄激素受体(N-20;圣克鲁茨)和p63(550025;Becton Dickson转导实验室)。
核磁共振成像
对单个小鼠进行MRI评估,以检测前列腺肿瘤。简而言之,用2%的异氟烷对小鼠进行麻醉,并在Bruker 4.7-T 40-cm孔磁铁上获得图像,该磁铁带有一个商业的7cm内径鸟笼线圈,类似于前面描述的方案19首先获得低分辨率矢状和轴向侦察图像,然后获得高空间分辨率图像T型2-加权轴向图像(重复间隔(T型R(右))=3800 ms,有效回波时间(T型E类)=35ms,每相位编码步8个回波,空间分辨率=1.0mm层厚×112μm×112μm,平面分辨率,重复4次数据采集,成像时间8-9分钟)。如前所述,通过Kaplan–Meier分析获得无侵袭和累积生存曲线19.
致谢
我们感谢T.Maeda和T.Jacks提出的有益建议;D.Peeper、C.Schmitt和M.Serrano交换未出版的数据并协调手稿的提交;N.Hay、U.Greber和S.Hemmi代表试剂;L.Cai和L.DiSantis对手稿进行批判性阅读和编辑;潘多尔菲实验室其他成员征求意见和讨论;分子细胞学核心设施的K.Manova和C.Farrell协助IHC分析;以及C.Le、C.Matei、D.Procissi和I.Buchanan进行MRI分析。这项工作得到了国家卫生研究院对P.P.P的拨款支持。
工具书类
-
1.Lowe SW,Cepero E,Evan G.内在肿瘤抑制。自然。 2004;432:307–315.doi:10.1038/nature03098。[内政部] [公共医学] [谷歌学者]
-
2塞拉诺·M,马萨诸塞州布拉斯科。衰老的压力。当前Opin细胞生物学。 2001;13:748–753.doi:10.1016/s0955-0674(00)00278-7。[内政部] [公共医学] [谷歌学者]
-
三。Campisi J.细胞衰老是一种肿瘤抑制机制。趋势细胞生物学。 2001;11:S27–S31。doi:10.1016/s0962-8924(01)02151-1。[内政部] [公共医学] [谷歌学者]
-
4Vogelstein B,Lane D,Levine AJ。浏览p53网络。自然。 2000;408:307–310.doi:10.1038/35042675。[内政部] [公共医学] [谷歌学者]
-
5Di Cristofano A,Pandolfi PP。PTEN在肿瘤抑制中的多重作用。单元格。 2000;100:387–390.doi:10.1016/s0092-8674(00)80674-1。[内政部] [公共医学] [谷歌学者]
-
6Stambolic V等。p53对PTEN转录的调节。分子细胞。 2001;8:317–325.doi:10.1016/s1097-2765(01)00323-9。[内政部] [公共医学] [谷歌学者]
-
7Mayo LD,Donner DB。磷脂酰肌醇3-激酶/Akt途径促进Mdm2从细胞质到细胞核的移位。美国国家科学院院刊,2001年;98:11598–11603.doi:10.1073/pnas.181181198。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
8Zhou BP等。HER-2/neu通过Akt介导的MDM2磷酸化诱导p53泛素化。自然细胞生物学。 2001;3:973–982.doi:10.1038/ncb1101-973。[内政部] [公共医学] [谷歌学者]
-
9Freeman DJ等。PTEN肿瘤抑制剂通过磷酸酶依赖和独立机制调节p53蛋白水平和活性。癌细胞。 2003;3:117–130.doi:10.1016/s1535-6108(03)00021-7。[内政部] [公共医学] [谷歌学者]
-
10Trotman LC、Pandolfi PP.PTEN和p53:谁会占上风?癌细胞。 2003;3:97–99.doi:10.1016/s1535-6108(03)00022-9。[内政部] [公共医学] [谷歌学者]
-
11Schmitt CA等人。由p53和p16INK4a控制的衰老程序有助于癌症治疗的结果。单元格。 2002;109:335–346.doi:10.1016/s0092-8674(02)00734-1。[内政部] [公共医学] [谷歌学者]
-
12Suzuki H等。多转移性前列腺癌组织中PTEN/MMAC1基因改变的局部异质性。1998年癌症研究;58:204–209.[公共医学] [谷歌学者]
-
13Feilotter HE、Nagai MA、Boag AH、Eng C、Mulligan LM。原发性前列腺癌中PTEN和10q23区的分析。致癌物。 1998;16:1743–1748.doi:10.1038/sj.onc.1200205。[内政部] [公共医学] [谷歌学者]
-
14Muller M,Rink K,Krause H,Miller K.前列腺癌中PTEN/MMAC1突变。前列腺癌前列腺疾病。 2000;3:S32。doi:10.1038/sj.pcan.4500457。[内政部] [公共医学] [谷歌学者]
-
15.Hermans KG等。PTEN基因座周围一个小区域的缺失是前列腺癌异种移植和细胞系中的主要10号染色体改变。基因染色体癌。 2004;39:171–184.doi:10.1002/gcc.10311。[内政部] [公共医学] [谷歌学者]
-
16Qian J等。T(2–3)N(1–3)M(0)期前列腺癌中p53缺失和c-myc过度表达是癌症进展的潜在标志。中度病理学。 2002;15:35–44.doi:10.1038/modpathol.3880487。[内政部] [公共医学] [谷歌学者]
-
17Navone NM等。前列腺癌骨转移中的p53突变表明,在原发部位选择的p53突变体定义了具有转移潜能的病灶。乌洛尔杂志。 1999;161:304–308.[公共医学] [谷歌学者]
-
18Di Cristofano A、De Acetis M、Koff A、Cordon-Cardo C、Pandolfi PP.Pten和p27KIP1协同抑制小鼠前列腺癌肿瘤。自然遗传学。 2001;27:222–224.doi:10.1038/84879。[内政部] [公共医学] [谷歌学者]
-
19Trotman LC等。铂剂量决定前列腺癌的进展。《公共科学图书馆·生物》。 2003;1:385–396.doi:10.1371/journal.pbio.0000059。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
20Jonkers J,Berns A.散发性癌症的条件性小鼠模型。《自然反击癌症》。 2002;2:251–265.doi:10.1038/nrc777。[内政部] [公共医学] [谷歌学者]
-
21Wu X,等。组织特异性基因消融前列腺上皮细胞特异性Cre转基因小鼠模型的建立。机械开发2001;101:61–69.doi:10.1016/s0925-4773(00)00551-7。[内政部] [公共医学] [谷歌学者]
-
22Dimri GP等。一种生物标记物,用于识别培养物和活体老化皮肤中的衰老人类细胞。美国国家科学院院刊1995;92:9363–9367.doi:10.1073/pnas.92.20.9363。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
23温伯格RA。基因、病毒和细胞玩的猫和老鼠游戏。单元格。 1997;88:573–575.doi:10.1016/s0092-8674(00)81897-8。[内政部] [公共医学] [谷歌学者]
-
24Miyauchi H等,Akt通过p53/p21依赖性途径负调节人类内皮细胞的体外寿命。EMBO J.2004;23:212–220.doi:10.1038/sj.emboj.7600045。[内政部] [PMC免费文章] [公共医学] [谷歌学者]
-
25Stewart SL、King JB、Thompson TD、Friedman C、Wingo PA。美国癌症死亡率监测,1990-2000年。MMWR监督总结。 2004;53:1–108.[公共医学] [谷歌学者]
-
26Levi F,Lucchini F,Negri E,Boyle P,La Vecchia C.西欧前列腺癌死亡率水平。前列腺。 2004;60:46–52.doi:10.1002/pros.20058。[内政部] [公共医学] [谷歌学者]
-
27阿巴特·申C,沈美美。前列腺癌的分子遗传学。基因开发2000;14:2410–2434.doi:10.1101/gad.819500。[内政部] [公共医学] [谷歌学者]
-
28Bykov VJ等。通过低分子量化合物恢复突变p53的肿瘤抑制功能。《自然医学》,2002年;8:282–288.doi:10.1038/nm0302-282。[内政部] [公共医学] [谷歌学者]
-
29Vassilev LT等。MDM2小分子拮抗剂对p53通路的体内激活。科学。 2004;303:844–848.doi:10.1126/science.1092472。[内政部] [公共医学] [谷歌学者]
-
30Maeda T等。原癌基因Pokemon在细胞转化和ARF抑制中的作用。自然。 2005;433:278–285.doi:10.1038/nature03203。[内政部] [公共医学] [谷歌学者]
关联数据
本节收集本文中包含的任何数据引用、数据可用性声明或补充材料。

