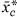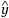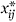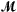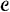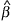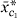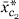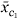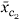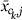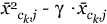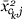树木采伐
作为我们的起点,我们有基因表达数据x个
ij公司
对于基因我= 1,2, ...第页和样品j个=1,2。。。n个和响应措施年= (年1,年2, ...年
n个
)对于每个样本(每个年
j个
可以是向量值)。反应度量可以有多种形式:例如,定量度量,如对治疗的反应百分比、删失生存时间或K(K)癌症课程。表达式数据x个
ij公司
可能来自cDNA微阵列,在这种情况下,它表示目标样品相对于参考样品的对数红绿比。或者x个
ij公司
可能是寡核苷酸阵列的表达水平。
基本方法由两部分组成:基因表达谱的层次聚类和响应模型。每个集群的平均表达式配置文件为响应模型提供了潜在的特征(输入)。
我们用以下公式表示基因簇X(X)
c(c)
,以及相应的平均表达式配置文件 c(c)
= (
c(c)
,1,
c(c)
,2, ...
c(c)
,个). 从开始第页基因,一个层次聚类将基因聚集在第页-1个后续步骤,直到所有基因都落入一个大簇。因此,它生成的总数为第页+ (第页-1)=2第页-1个簇,我们表示为c(c)1,c(c)2, ...c(c)2第页- 1.
c(c)
= (
c(c)
,1,
c(c)
,2, ...
c(c)
,个). 从开始第页基因,一个层次聚类将基因聚集在第页-1个后续步骤,直到所有基因都落入一个大簇。因此,它生成的总数为第页+ (第页-1)=2第页-1个簇,我们表示为c(c)1,c(c)2, ...c(c)2第页- 1.
反应模型通过一些平均基因表达谱及其产物来近似反应测量,具有捕获加性和相互作用效应的潜力。为了便于构建交互模型,我们将每个x个
ij公司
样本的最小值为0:
符号
表示集群的平均表达式配置文件c(c),使用这些转换值。翻译只是为了使模型中的交互更容易理解。请注意,集群中使用了未翻译的值。
对于定量反应年
j个
,j个= 1,2,...n个,模型采用以下形式:
其中β
k个
和β
kk’
是通过最小化平方误差之和∑估计的参数
j个
(年
j个
-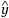 j个
)2.作为每个
j个
)2.作为每个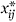 最小值为0,产品术语表示相关基因之间的正协同或负协同。
最小值为0,产品术语表示相关基因之间的正协同或负协同。
显然,将所有簇都包括在方程式的总和中是不可行的,甚至是不可取的2相反,我们以如下方式逐步建立模型。最初是模型中的唯一术语
是常数函数1。候选人条件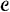 包括2个第页-1个平均表达谱
包括2个第页-1个平均表达谱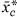 。在每个阶段,我们都考虑由以下术语组成的所有产品
。在每个阶段,我们都考虑由以下术语组成的所有产品 和中的一个术语,并添加在得分统计方面最能改善模型拟合度的术语S公司。我们将继续执行,直到达到最大条款数已添加到模型中。
和中的一个术语,并添加在得分统计方面最能改善模型拟合度的术语S公司。我们将继续执行,直到达到最大条款数已添加到模型中。
例如,在第一阶段,我们输入最佳平均表达式配置文件
; 这对应于常数函数1。生成的模型具有以下形式
j个
= 0+1
0+1 ,j个,其中0,1用最小二乘法求出。在第二阶段,模型中可能添加的内容如下2
,j个,其中0,1用最小二乘法求出。在第二阶段,模型中可能添加的内容如下2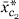 ,j个或12,j,j个对于某些群集c(c)2.
,j个或12,j,j个对于某些群集c(c)2.
通常,该算法可以生成包含三个或更多平均表达式配置文件乘积的项。但是用户可以对交互的顺序进行明确的限制,我,模型中允许。为了简化解释,在这里的示例中,我们设置了我=2,表示产品仅限于成对产品。这是通过只考虑单一术语(非产品)来实现的作为第二步的候选人。
方程中具有两两相互作用的模型2通常用于统计应用程序。相互作用通常在包含重要的附加项之后,以临时的方式包含在内。Friedman的MARS(多元加性回归样条)程序是一个例外[三]. 这是一种通用的自适应学习方法,它将交互模型建立为输入的分段线性函数的乘积。该模型的建立方式与树木收获过程相同。MARS是一种非常流行的方法,并激发了本文中的一些想法。
有一些关键的计算细节,使得该算法能够在实际应用中快速运行。首先,在开始正向逐步过程之前,我们需要所有2个变量的平均表达式配置文件第页-1个集群。这是通过使用层次聚类后可用的树结构以自然递归方式实现的:节点中的平均表达式配置文件是子节点两个平均配置文件的加权平均值,其中权重是子节点的大小。其他特定于节点的统计信息(如方差和方差内)可以用类似的方法计算。
其次,在算法的第二步中,我们必须搜索所有2个第页-1聚类,以找到最能提高模型拟合度的项。这是通过将候选平均表达式轮廓与模型中已有的术语正交,然后计算每个候选术语的得分测试来实现的。通过定量响应和最小二乘法,这个过程准确地给出了每个候选项对模型的贡献。对于生存、分类和其他类似的基于hood的模型,它是一种广泛使用的近似值。
其他功能和问题
数据规范化
与大多数微阵列实验集一样,每个实验的数据来自不同的芯片,因此必须首先进行标准化,以考虑芯片的变化。我们假设每个实验的值j个已经居中,也就是说x个
ij公司
→x个
ij公司
- (1/第页) ∑
我
x个
ij公司
.
聚类方法和准则的选择
树收获过程只是从一组集群开始,这些集群可以由任何集群方法提供。由于微阵列数据的普及性和有效性,我们选择了基于层次聚类的程序(例如,参见[2])。簇集以层次方式方便地排列,并相互嵌套。具体来说,如果聚类树在两个不同的级别上被截断,分别生成四个和五个簇,那么四个簇将嵌套在五个簇中。因此,可以同时查看所有不同分辨率的聚类。此功能便于解释树收获结果,并且不是大多数其他聚类方法的特性。尽管如此,其他聚类方法可能被证明在树收获过程中具有优势,包括K-means聚类、自组织映射[4],以及允许重叠簇的程序(例如,基因剃毛[1])。聚类标准的选择也会影响结果。我们再次关注艾森等. [2]并采用平均连锁聚类,应用于基因的相关矩阵。相关性的使用使得聚类对单个基因的缩放不变性。扩展最终的聚类(见下文)可以减轻结果对聚类方法和标准选择的敏感性。
偏向较大的簇
典型的基因表达数据集有许多高度相关的基因。此外,收获过程中考虑的大多数集群都是其他集群的子集。因此,如果平均表达式配置文件
在程序的第2步中发现,最能改善模型的拟合度,很可能某些较大簇(可能包含所选簇)的平均表达谱几乎与在其他条件相同的情况下,我们更喜欢较大的星团,因为它们更有可能具有生物学意义。大簇可能是由参与生物过程的基因通路或包含不同细胞类型的异质实验样品引起的。此外,与结果相关的大集群的发现比小集群的发现不太可能是虚假的,因为小集群比大集群多得多。由于这些原因,我们将选择过程偏向于较大的簇。具体来说,如果集群c的得分为S公司
c(c)
,我们选择了最大的集群c(c)'谁的分数S公司
c(c)
'在最佳因子(1-α)范围内,这是令人满意的S公司
c(c)
'≥ (1 - α)S公司
c(c)
.参数α可由用户选择:我们在示例中选择了α=0.10。集群c(c)'通常包含c(c),但这不是必需的。虽然这会使选择偏向于更大的簇,但如果单个基因的贡献是惊人和独特的,那么它仍然可以被选择。
模型尺寸选择和交叉验证
建立了一个包含大量术语的收获模型,M(M),我们执行向后删除,在每个阶段丢弃导致平方和最小增加的项。我们继续,直到模型只包含常数项。这给出了一系列具有1、2、……个项的模型。。。M(M),我们希望选择模型大小,因此选择其中一个模型。模型尺寸由以下方式选择K(K)-折叠交叉验证。数据被拆分为K(K)部分。对于每个k个= 1,2, ...K(K)除了k个第个部分,然后是k个该部分是根据训练好的模型进行预测的。计算结果的平均值k个= 1,2, ...K(K)。这将在接下来两部分的示例中进行说明。
扩展集群
层次聚类使用基因的离散分区序列。因此,对于给定的簇,可能有不在该簇中的基因与簇的平均表达谱的相关性比簇中的某些基因更高。为了解释这一点,我们只需在最后一组簇中寻找此类基因,并将其作为属于每个簇的“额外基因”进行报告。
我们总结了算法1(方框1)
一般响应变量的树木收获
树收获方法可以应用于最常见的响应数据类型。给定的响应年=(年1,年2, ...年
n个
),我们形成了基于模型的近似η=(η1, η2, ... η
n个
)要最小化损失函数:
每个数量η
j个
是平均基因表达谱的函数,其形式如方程式所示2:
表中列出了一些常见的响应类型和损失函数1.
如前一节所述,该模型是以向前逐步的方式建立的。考虑到
是参数β={β的函数
k个
,β
k、 k’
},将每个新术语添加到模型中是基于分数统计的大小:
和类似的β
k、 k’
截尾生存时间和分类反应模型将在接下来的两部分中进行说明。
淋巴瘤患者的生存率
图1显示了本例中使用的数据集,包括36例弥漫性大细胞淋巴瘤(DLCL)患者的3624个基因表达测量值。Alizadeh中描述了这些数据等. [5]. 列标签表示不同的患者,行标签标识基因。我们对基因进行了层次聚类,对样本进行了单独聚类。每个聚类都会产生一个(非唯一的)排序,确保相应树状图的分支不会交叉。图1显示原始数据,并相应地对行和列进行排序。
对于36名患者中的每一位,都有一个(可能经过审查的)生存时间;这一时间段为1.3至102.4个月,36例患者中有19例在研究期间死亡。一个合适的反应模型是考克斯的比例风险模型[6]. 其形式如下:
在这里z(z)
j个
= (z(z)1j个,z(z)2j个, ...z(z)
mj(百万焦耳)
)是米样本的风险因素(特征)j个、和小时(t吨|z(z)
j个
)表示具有特征值的个人的危险函数z(z);小时0(t吨)是具有风险因素的个人的基线风险函数z(z)=0。未知函数第页(z(z)
j个
)表示随时死亡的对数相关风险t吨对于个人z(z)=z(z)
j个
与个人相比z(z)= 0. 在树木收获模型中(z(z)1j个,z(z)2j个, ...z(z)
mj(百万焦耳)
)是平均表达谱,我们取第页(z(z)
j个
)应采用以下形式:
如方程式所示2。树收获算法根据部分似然计算近似得分测试,以决定在每个阶段输入哪个术语。
我们运行了最多允许六个术语的收获过程,结果如表所示2.
需要一些解释。在每个阶段,“节点”指的是选择其平均表达式配置文件添加到模型中的集群“Parent”是模型中已有的簇数,即乘以节点平均表达式配置文件;Parent=0表示常量函数1。Node或Parent以“s”开头的节点表示单个基因分数”是指通过添加术语获得的分数值;它大致是高斯变量,因此≥2的值相当大。
只关注第一个集群的选择,图2显示所有聚类得分。绿色水平线绘制在(1-α)乘以最大得分(α=0.1)处,我们选择了该线上方最大的簇(蓝色点)。该簇是八烯簇3005,如图所示三.
总的来说,生成的模型具有以下形式:
正系数表示风险增加。训练集和交叉验证曲线如图所示4交叉验证(CV)曲线的最小值出现在一个术语中,这表明后续术语可能无法改善预测。
基因簇如图所示三和列在本文联机版本提供的附加数据文件中。仅关注第一个聚类(3005),我们计算了36名患者的平均表达。然后将患者分为两组:平均表达低于中位数的患者(第1组)和平均表达高于中位数的人群(第2组)。这两组的Kaplan-Meier生存曲线如图所示5和明显不同(第页=2.4 x 10-5).
如果3624个基因中的每个基因在Cox得分统计中从最低(1)到最高(3624)排序,那么集群3005中8个基因的平均排名为3574.5。因此,这些基因是预测存活率的最强基因之一,但不是最强的八个基因。相反,它们是一组表达谱非常相似的基因,与生存高度相关。
人类肿瘤数据
在这个例子中,响应是一个指定癌症类别的分类变量。我们使用Ross描述的61个肿瘤的子集等. [7]和谢尔夫等. [8]省略了两个前列腺肿瘤和一个未知类别。每个肿瘤都有6830个基因的表达值,其在不同癌症类别中的分布如表所示三.
在这里,树收获方法以逐步的方式建立了一个多元逻辑回归(MLR)模型,使用与Cox生存数据模型相似的步骤。这里的目标是在给定表达式值的情况下,对肿瘤类别的概率进行建模。一般来说,如果类变量由年取{1,2,…,中的值。。。,J型}预测变量由x个1,x个2, ...,x个
第页
线性MLR模型的形式如下:
和以前一样x个1将是聚类平均值,可能是单个基因,或这些基因的成对产物。逻辑变换是一个自然的尺度,可以在这个尺度上对K(K)概率;逆变换:
保证概率总和为1并且是正的。该模型通常采用多项式极大似然法进行拟合。因为反应确实是多层面的,我们不希望出现单一的x个能够区分所有癌症类别;这意味着单个基因的平均值会产生一个区分癌症类别的顺序。通常需要几个。
在每个阶段,树收获算法都会考虑用一个新的术语来扩充当前拟合的MLR模型,候选项是节点平均值、单个基因或模型中已有术语的这些项的乘积。如前所述,使用了适合多项式模型的得分统计。
表中显示了允许七个术语的树木收获拟合结果4偏差是对多项式模型缺乏f的度量,我们看到模型中有七个项,我们有一个饱和拟合(该模型对每个观测值和相关类别产生的概率估计基本上为1)。这几乎肯定是一种飞越情况,因为我们将56个参数拟合到61个观测值。
图6显示了模型发现的七个术语中的所有基因;任意选择列顺序来分隔癌症类(并在癌症类中随机选择)。我们使用十倍交叉验证来为模型找到大量术语。图7显示了根据偏差统计(-2×log-likelihood)得出的结果。对于这些数据,两项模型将CV偏差曲线最小化,并对应于图中的顶部两个带6.
图8显示了前两个簇中每个簇的平均表达散点图,样本按癌症类别识别。癌症类别中存在明显的分离。
仿真
我们进行了一项模拟实验,以评估树木采伐对“真实”结构的发现程度。为了确保基因表达测量在大小和相关性上是真实的,我们使用了3624×36个淋巴瘤表达测量的矩阵来进行我们的研究。然后生成人工生存和审查时间,以生成用于收获的模拟数据集。
考虑了两种情况,相加和相互作用。对于加性场景,我们随机选择了一个簇,并生成了相对风险为2的删失生存时间,作为其平均表达谱的函数。如表所示5随机选择的聚类来自单个基因、小簇(<10个基因)或大簇(介于10到300个基因之间)。采伐树木只允许进入一个学期。
对于交互场景,我们随机选择了一个集群c(c)1有两到十个基因,然后选择第二个簇c(c)2是包含两到十个基因的簇,其平均表达谱与c(c)1。这使得两个集群尽可能独立,使收获过程有最大机会发现它们的相互作用。然后用相对风险函数4生成生存数据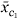 + 4
+ 4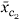 + 3[-第页]其中第页是的投影在和树木采伐被允许进入三个阶段。
+ 3[-第页]其中第页是的投影在和树木采伐被允许进入三个阶段。
结果显示在表的顶部面板中5。这些数字是五次模拟的平均值。这些列显示了真实簇中的平均基因数、通过树木采伐发现的簇中平均基因数以及通过树木采收发现的基因在真实簇中所占的比例,反之亦然。最后一列显示了真实簇的平均表达谱与估计簇的平均绝对相关性。对于交互作用场景,这些数量是指组成交互作用的基因集合。如果发现多个相互作用,则报告与真实相互作用簇重叠最大的一个。我们看到,当真正的簇是单个基因时,树木采伐返回的簇有点太大,而当真正的集群很大时,返回的簇又太小。在加性场景中,它在发现真正的簇或与之类似的簇方面做得相当好。然而,它只正确地发现了大约四分之一的交互。需要更多的样本才能准确地找到这么多基因之间的相互作用。另一方面,最右边一列中的相关性都很高,这表明树木采伐能够找到与真实集群几乎一样好的集群。
桌子的中间面板5显示了相对风险降至1.0时附加场景的结果。正如预期的那样,尽管平均相关性仍在0.60左右,但情况有所恶化。
为了研究更多的样本是否会改善相互作用的检测,我们将相同的方法应用于一组129个样本和1622个基因,这些样本来自一项尚未发表的乳腺癌研究(T.Sorlie、C.Perou和合作者,个人通信)。与之前一样,我们使用了表达式值和模拟的合成生存时间集。结果显示在表的底部面板中5现在,树收获过程很好地恢复了相互作用。样本数量越多,加上基因数量越少,性能就会显著提高。
非线性树木收获模型
在上述收获过程中,基因表达的影响是线性建模的。因此,在对每个术语建模时,我们假设增加或减少基因表达对结果具有一致的影响。然而,基因具有非线性效应在生物学上是合理的:例如,增加表达可能与更长的生存期相关,但仅在一定程度上相关。超过这个水平,可能会导致相同或更差的生存率。
为了考虑到非线性效应,每个基因可以使用灵活的功能基础。然而,由于有大量的基因,这往往会很快过盈。因此,我们允许每个基因使用简单的二次函数:
我们首先正交化b条(x个)相对于同一基因的线性项,然后允许转换的表达b条(x个)代替表达式x个在树收获模型中。具体而言,该模型的形式与方程式相同2:
哪里
等于任一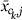 或
或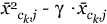 选择γ来制造
选择γ来制造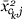 与不相关在数据集上。
与不相关在数据集上。
如果一个二次项乘以一个正系数,那么一个基因的效应是“U”型的,先减小后增大。对于负系数,效果是一个倒“U”。两个二次项之间的产物相互作用表明两个基因之间存在强烈的协同效应,而忽略了表达方向(低于或高于平均值)。当非线性选项用于收获时,程序在每个阶段尝试线性和非线性项,并选择得分最高的项。
淋巴瘤数据续
我们尝试使用淋巴瘤数据集的非线性选项进行树木采伐,它给出了表中所示的前四个术语6.第2-4项中输入了二次项;与之前的线性模型拟合相比,这些模型拟合到第3项的效果更好,但之后的拟合效果并不好。该模型中的集群如图所示9.
例如,在第二个集群中(图中标记为“2”9)我们发现,中等表达水平的存活时间最长,而非常低或非常高表达水平的生存时间更差。
总体而言,与线性模型相比,非线性模型缺乏显著的改进,这使得人们更相信每个项的线性形状在本例中是合适的。然而,二次模型很可能对其他基因表达实验有用。