摘要
1简介
Maiden介绍了多焦点序列分型(MLST)等。(1998)作为一种从特定物种中鉴定细菌分离物的方法。它依赖于几个400–500 bp的家政基因片段的测序来确定分离物的类型。MLST最初被提议用于脑膜炎奈瑟菌(少女等。,1998),但已应用于50多个不同物种(Maiden,2006; 乌文和少女,2003). MLST结果可以在实验室之间轻松共享和比较(Urwin和Maiden,2003),并定期在上提供http://pubMLST.org/网站由牛津大学主办网址:http://www.mlst.net/帝国理工学院和http://web.mpiib-berlin.mpg.de/mlst/马克斯·普朗克研究所主办的网站。
在中性模型下模拟MLST的能力有助于对采样数据集进行解释,例如推断进化参数的值(Fearnhead等。,2005; 弗雷泽等。,2005),分析选择所扮演的角色(巴基等。,2008),以应用近似贝叶斯计算方法(Marjoram等。,2003; 威尔逊等。,2009)或者测试家谱推断的方法(迪德洛和法鲁什,2007; 法卢什等。,2006; 特纳等。,2007). 对于最后一项任务,有必要模拟克隆系谱(Guttman,1997)这就产生了数据以及数据本身。
之前已经描述了MLST仿真的几种方法。两位弗雷泽等。(2005)和Falush等。(2006)使用了一种正向时间方法,需要模拟整个群体(而不仅仅是样本),并等待达到平衡。羽毛头等。(2005)使用了向后时间(合并)模拟程序MS(Hudson,2002)生成一个包含MLST基因座的单个大遗传区域,该区域彼此之间的距离很大(10 kb)。在这里,我们提出了一种更有效的方法来模拟MLST数据。
2型号
我们假设的基本模型是结合基因转化(Wiuf和Hein,2000). 该模型类似于流行的结合重组模型(Hudson,1983),但假设当两个细胞重组时,产生的基因组与受体的基因组相同,除了来自供体的(小)相邻片段。由于这是细菌重组的方式(无论是通过接合、转化还是转导),因此结合基因转化是模拟MLST数据的合适模型。我们跟随维夫和海因(2000)假设基因转换事件同样可能在基因组上的任何一点启动,并且输入的长度以参数δ的几何分布,这与经验证据(Falush等。,2001; 羽毛头等。,2005; 乔利等。,2005).
3算法
维夫和海因(2000)提出了一种结合基因转换模拟单个位点的算法。这里我们从三个方面扩展了他们的算法。首先,我们使用Didelot和Falush的结果进行多轨迹模拟(2007,式4),重组的起点是δ倍,更可能位于一个位点的起点,而不是位于该位点内的某个位点。其次,我们只模拟祖先的材料(哈德逊,1983;2002)为了提高效率,每个谱系中至少有一个个体的祖先位置。我们使用拒绝采样来忽略任何不会将谱系祖先物质分裂为两个非空子集的重组事件。第三,我们联合模拟克隆系谱(Guttman,1997)使用数据。克隆系谱是通过追踪每个重组事件中受体的谱系获得的。克隆谱系学的正确模拟需要允许它不携带任何祖先物质,不像上面描述的其他谱系。
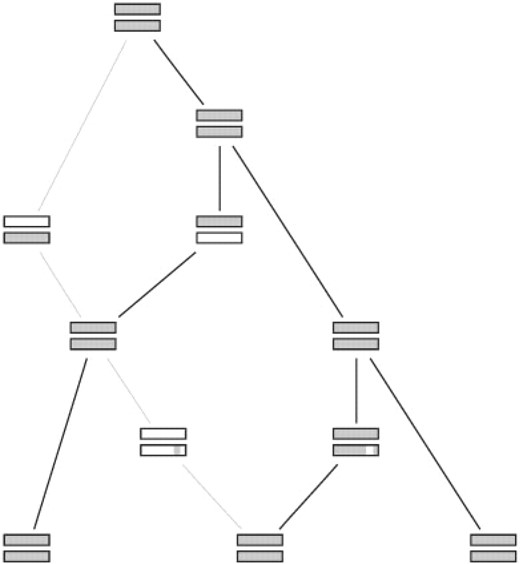
图1说明了我们的算法对三个分离物和两个基因样本的工作。样本的祖先可以追溯到时间上,直到所有分离株在所有位点找到共同的祖先。克隆谱系以粗体表示图1第二分离物是重组的结果,其中第二基因的片段被导入,并且第一分离物的第一基因从克隆根上方导入。重组使不同的基因片段具有不同的系谱:尽管分离物2和3在第一个基因中关系最密切,但分离物1和2在第二个基因的插入片段中关系最紧密。
图1。
三个分离株和两个基因座的模拟样本的系谱史。每个基因座都由一个方框表示,其中祖先的物质是灰色的。克隆谱系以粗体显示。
我们的算法通过对祖先图(Griffiths和Tavare,1994)我们允许通过使用与MS(Hudson,2002). 然后通过将突变作为泊松过程添加到祖先图中来生成数据。我们使用Jukes和Cantor的突变模型(1969)默认情况下,但我们的程序可以与seq-gen(Rambaut和Grass,1997)以模拟各种其他模型。
4结论
SimMLST联合模拟MLST数据和分离物之间的克隆关系。它以灵活的扩展多方位对齐(XMFA)格式输出MLST数据,以Newick格式输出克隆谱系。SimMLST还可以与图形绘制软件包DOT(Gansner等。,1993)表示样本的完整系谱历史(如图1).
SimMLST比以前的方法更有效,因为它只模拟对数据有影响的重组事件,而不是那些从测序区域或谱系祖先物质中脱落的重组事件。因此,它生成用于模拟数据的祖先图的大小是最佳的。与所有基于聚合的方法一样,SimMLST的时间和内存需求的增长速度远远快于总体重组率ρ的线性增长速度。然而,它可以支持高达数千的ρ值,这比任何细菌物种(Didelot和Falush,2007; 羽毛头等。,2005; 弗雷泽等。,2005; 乔利等。,2005).
SimMLST的高效性有助于从模拟中推断进化参数,这通常需要生成数千个具有广泛参数的数据集。还需要生成比MLST更大的数据集(在测序位点的数量上),其中ρ将更高。假设与MLST数据中观察到的复合率相似脑膜炎球菌SimMLST可以生成多达数百千个碱基对的数据集。然而,为了模拟频繁重组物种的整个基因组,需要近似于结合与基因转化过程。
致谢
作者感谢编辑和两位匿名审稿人的深刻评论。
基金:威康信托。
利益冲突:未声明。
参考文献
等选择在脑膜炎奈瑟菌谱系形成和毒力进化中的作用
, 程序。美国国家科学院。科学。美国
, 2008
,卷。 105
(第15082
-15087
) , . 利用多点序列数据推断细菌的微进化
, 遗传学
, 2007
,卷。 175
(第1251
-1266
) 等长期胃定植过程中的重组和突变幽门螺杆菌:时钟频率、重组大小和最小年龄的估计
, 程序。美国国家科学院。科学。美国
, 2001
,卷。 98
(第15056
-15061
) 等沙门氏菌错配诱导的物种形成:模型和数据
, 菲洛斯。事务处理。R.Soc.B生物。科学。
, 2006
,卷。 361
(第2045
-2053
) 等中的重组分析空肠弯曲菌来自MLST人口数据
, 《分子进化杂志》。
, 2005
,卷。 61
(第333
-340
) 等细菌病原菌的中性微流行演变
, 程序。美国国家科学院。科学。美国
, 2005
,卷。 102
(第1968
-1973
) 等有向图的一种绘制技术
, IEEE传输。柔和。工程师。
, 1993
,卷。 19
(第214
-230
) , . 可变环境中中性等位基因的抽样理论
, 菲洛斯。事务处理。R.Soc.B生物。科学。
, 1994
,卷。 344
(第403
-410
) . 大肠杆菌自然群体的重组和克隆性
, 经济趋势。进化。
, 1997
,卷。 12
(第16
-22
) . 基因内重组中性等位基因模型的性质
, 西奥。大众。生物。
, 1983
,卷。 23
(第183
-201
) . 在Wright-Fisher中性遗传变异模型下生成样本
, 生物信息学
, 2002
,卷。 18
(第337
-338
) 等突变、重组、种群历史和选择对遗传多样性模式的影响脑膜炎奈瑟菌
, 分子生物学。进化。
, 2005
,卷。 22
(第562
-569
) , . . 蛋白质分子的进化
, 哺乳动物蛋白质代谢。
, 1969
纽约州纽约市
学术出版社
(第21
-132
) 等多焦点序列分型:一种识别致病微生物群体克隆的便携式方法
, 程序。美国国家科学院。科学。美国
, 1998
,卷。 95
(第3140
-3145
) . 细菌的多焦点序列分型
, 每年。微生物评论。
, 2006
,卷。 60
(第561
-588
) 等无概率马尔可夫链蒙特卡罗
, 程序。美国国家科学院。科学。美国
, 2003
,卷。 100
(第15324
-15328
) , . Seq-Gen:DNA序列沿系统发育树演化的蒙特卡罗模拟应用
, 生物信息学
, 1997
,卷。 13
(第235
-238
) 等使用已知血统的模拟人群评估eburst的可靠性
, BMC微生物。
, 2007
,卷。 7
第页。 30
, . 多焦点序列分型:全球流行病学工具
, 微生物趋势
, 2003
,卷。 11
(第479
-487
) 等胃肠道病原体空肠弯曲菌的快速进化及重组的重要性
, 分子生物学。进化。
, 2009
,卷。 26
(第385
-397
) , . 与基因转化结合
, 遗传学
, 2000
,卷。 155
(第451
-462
)
作者注释
©作者2009。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oxfordjournals/org








{kind=link}