作者:Mark Archibald。
在上一篇文章中(ChemSpider幕后)我们讨论了在维护世界上最大的化学数据库之一的数据质量方面的一些挑战。我们将自动过滤确定为处理人类无法合理处理的大量记录时的关键工具。在这篇文章中,我们将更详细地介绍过滤的工作原理、面临的挑战以及人工干预所起的作用。
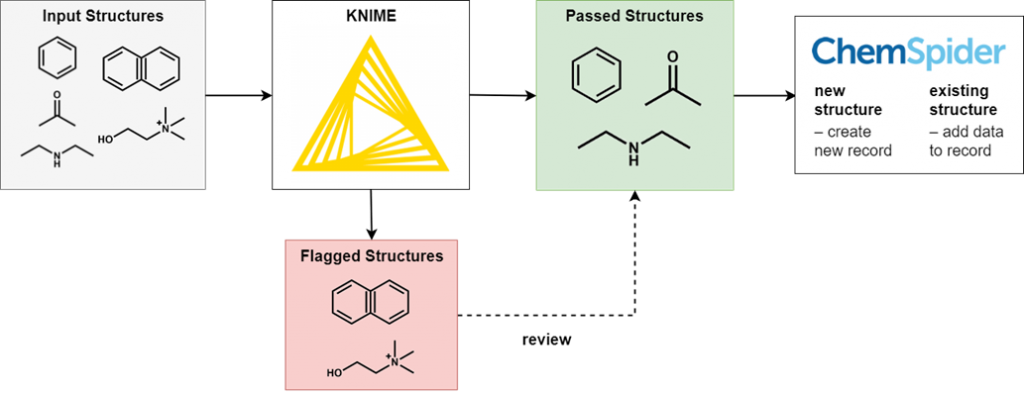
为了执行此过滤,我们使用KNIME,一个开源数据处理平台。由活跃的化学信息学社区开发的广泛的KNIME节点允许我们对我们处理的数据提出特定于化学的问题。简单地说,符合我们标准的输入化学结构被传递到下一个节点,而那些不符合标准的则被写入错误文件。处理完所有结构后,结果是一个成功通过所有过滤器的结构文件,以及由于各种原因被拒绝的几个(通常较小)结构文件。
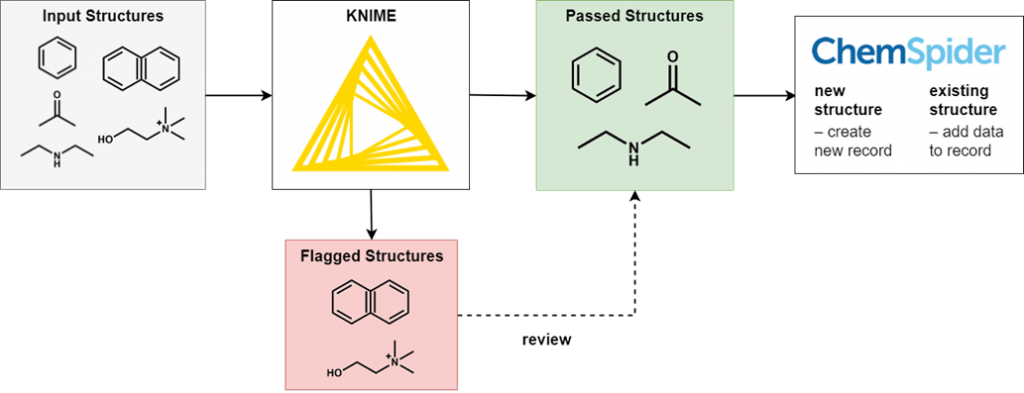
不可能完整地检查所有生成的文件,因为这将消除自动处理的省时优势。然而,所有类型的输出文件都会进行抽查,以确保准确性,并反复改进过滤标准。某些输出文件很可能出现误报,因此我们会对其进行全面检查。
格式和标识符
提交的文件可以是几种不同格式中的一种。最常见的是SDF(结构数据文件,一种包含多个结构和相关数据字段的化学结构格式)。这种格式的优点是它包含二维或三维结构,因此我们可以立即开始处理文件,而无需将标识符转换为结构。这意味着我们存放的最终结构更有可能与原始结构完全匹配。SDF格式的缺点是它是专门化的——许多用户可能不熟悉它,或者没有创建和显示文件的软件。
我们还收到不同的电子表格格式(excel、csv、tsv),其结构编码在基于文本的符号系统中,如微笑或InChI公司这种格式的优点是不需要专门的软件(前提是提交者对化合物使用SMILES或InChI)。缺点是结构需要在加工和沉积到ChemSpider之前转换为SDF。此外,这些格式包含有关原子及其连接性的信息,但缺少布局信息。这可能会引入错误,因为不同的结构绘图包可能会以稍微不同的方式解析这些结构,从而导致最终沉积结构的更改。
筛选条件
根据我们的经验和化学知识,我们判断化学结构的标准是明确的化学规则和不太明确的“经验法则”的混合。下面是这两者的示例。
空结构、查询原子和不正确的价
第一个过滤器是最简单的——ChemSpider是一个以结构为中心的数据库,因此不可能存放任何缺少结构的输入条目。
类似地,每个ChemSpider记录都需要一个单独定义的化学结构,因此我们排除了使用查询原子来表示变量原子或连接点的任何内容。
另一个简单的过滤器是排除原子具有无效价的结构。
充电不平衡
一般来说,ChemSpider中的条目应表示真实的、可隔离的化合物。这意味着我们过滤出总电荷不为零的结构。然而,我们对某些情况例外,在这些情况下,反离子通常不重要,只考虑带电物种是有用的,例如胆碱(ChemSpider记录).
包含未定义立体中心的结构
未定义的立体中心本身并不代表化学错误。然而,像下面所示的结构(胆固醇没有任何定义的立体中心)经常出现,尽管化学上是有效的,但它们不太可能代表预期的结构。
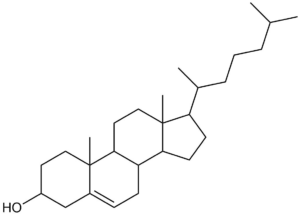
胆固醇骨架无立体化学
因此,我们有一个经验法则,排除包含两个以上未定义立体中心的结构。这不是一个硬性规定,而是试图在排除上述结构和包含未定义立体中心是有意且正确的结构之间取得平衡。
未定义立体中心的计数(通过检查InChI确定)有时包括常规排除立体化学楔的情况。例如,磷酸基团上没有楔形物的核酸和没有明确立体化学的金刚烷基团-用楔形物绘制这些化合物是不常见的,用户在搜索时很少使用楔形物。这些潜在的假阳性被筛选出来并手动审查。然后,馆长可以决定是否将它们包括在沉积中,从而提高过滤器的整体精度。
包含许多组件的结构
这是另一条经验法则——一种正确描述的化学物质可以含有多少独立成分没有上限。然而,根据经验,我们发现,排除具有四个以上独立组件的结构会删除最明显的无意义条目(例如。 试图描绘合金)同时保留大多数正确条目。
应用这一规则时,药物分子是误报的主要来源,因为它们通常是多水合物和/或具有多个反离子的盐(例如。盐酸伊立替康三水合物)。排除的水合物结构或含有普通药用盐的结构被标记为供人类审查。
同义词过滤器
此过滤器将指定给给定结构的同义词与其分子公式进行比较,并执行一些“常识”检查。例如,一个相对常见的错误是关联salt表单的名称(例如盐酸莫扎瓦普坦)带有自由底座的结构(莫扎瓦普坦)。在这种情况下,过滤器会删除包含“盐酸盐”的同义词,因为分子式不包含Cl。
智能
智能(维基百科页面)是描述一般化学结构的一种方式。它以SMILES为基础,但具有其他功能,允许指定可变链长、键数、氢数、可变键序或一个位置的多个潜在元素。
我们使用SMARTS来识别结构中常见的错误特征。其中包括:
- 五价氮描绘的叠氮化合物和重氮基团
- 与主体结构无关的“浮动”烷烃(可能是由于绘图程序中的意外点击引起的)
- 金属羧酸盐,描述为具有元素金属原子的质子化羧酸
- 六氟磷酸盐(和类似物种)被描述为五氟化磷和一个单独的氟离子
SMIRKS公司
SMIRKS是SMILES的进一步扩展,用于描述反应。我们不使用它来表示真实的反应,而是定义结构转换——允许我们修复简单的结构错误,这些错误可以通过破坏和创建键来解决。
一个例子是连接电荷分离的格氏试剂以提供更准确的描述:

重新连接Grignard
有机计量学
以机器可读格式编码有机金属结构的困难已被充分记录(化学杂志。信息模型。51, 12, 3149-3157)。有一个正在进行的IUPAC项目扩展InChI的功能但就目前而言,挑战依然存在。
每个ChemSpider记录基本上都基于InChI,因此我们受到当前限制的约束。这意味着我们不能描述配位键或非整数级键——任何绘制的键都被解释为标准共价键,每个原子贡献一个电子。
虽然我们通常不能以人类化学家喜欢的方式来表示有机金属结构,但我们仍然试图从各种可能的折衷中选择“错误最小”的结构。
二茂铁是这个问题的一个典型例子,说明了我们必须考虑的几个问题。下面显示了绘制二茂铁的几种常见方法(还有更多)。
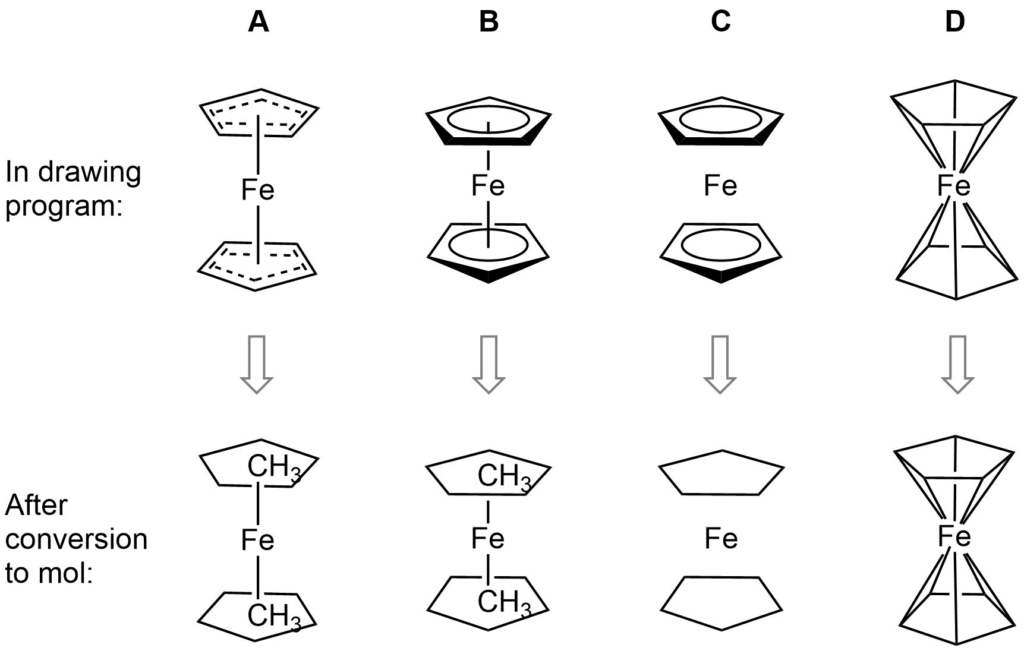
将二茂铁结构转换为摩尔格式可能会导致分子式、键序或价的错误
所示的大多数结构都利用了化学绘图包的扩展功能,以一种对人类化学家有吸引力且易于理解的方式来表示二茂铁的键合。不幸的是,一旦转换为简化但通用的mol格式,其中一些特性就会丢失,导致无意义的结构。虽然结构D没有改变,但这种表征还有其他问题:铁的化合价不正确,也不能表征环戊二烯配体的芳香性。
我们可以用有限的方法来描述ChemSpider中的二茂铁及其相关结构,但没有一种方法能够准确地表示键合,也没有一种观点能够满足无机化学家的要求。然而,我们可以从可能的折衷中选择“最不坏的”,并允许机器可读性:
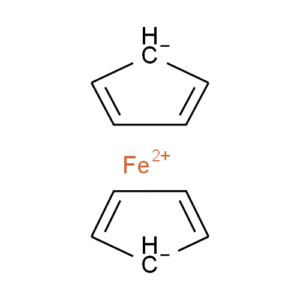
我们的妥协
尽管这种结构(ChemSpider记录)没有捕获二茂铁的触觉,而且单个碳上的电荷定位不准确,它保留了正确的总电荷和价,并且没有显示配体是sigma-键合的。
更广泛地说,我们应用一些规则和转换来标准化有机金属结构的表示。其中许多规则涉及根据金属和配体的性质,选择将金属-碳(或金属-杂原子)描述为共价还是离子。同样,在机器可读结构的限制范围内工作时,有必要做出妥协,但我们试图将“更离子”和“更共价”键分类。以下是一些示例:
- 从第1组和第2组金属上断开氧气
- 将氧气连接到所有其他金属
- 从钠、钾和钙中分离碳
- 将碳连接到第11组和第12组金属、p块金属和一些类金属
正如预期的那样,像这样的一般规则在某些情况下会失败。因此,我们有额外的、更具体的规则来覆盖例外情况,我们会反复完善这些规则。
但这些错误仍然出现在ChemSpider中!
目前,所述过滤仅适用于进入ChemSpider的新数据。多年来建立的完整ChemSpider数据库当然包含了这里描述的每个错误的示例。为了修复这些遗留错误,我们打算通过相同的质量过滤器运行整个数据库。这是一项重要的任务,有一些特定的挑战:需要人工审查的文件变得更大,处理时间和内存/CPU开销很高,数据集越大,出现误报的可能性越大。为了应对这些挑战,我们正在花时间完善新沉积的工艺,并通过过滤器运行完整ChemSpider数据库的子集定期检查我们的进度。我们知道您需要访问您可以信任的数据,所以我们希望确保这一点是正确的。随着项目的进展,我们将继续向您更新,敬请期待!