摘要
肌营养不良蛋白的零突变导致Duchenne型肌营养不良症(DMD)的致命病理,其中骨骼肌和心肌的病理进展。DMD患者死亡的很大一部分归因于与心室纤维化、心律失常和传导异常相关的心脏功能障碍,尽管肌营养不良蛋白突变与心脏缺陷之间的关系尚不清楚。在这里,我们测试了肌营养不良蛋白缺乏mdx小鼠的心脏病理是否可以通过心肌一氧化氮(NO)生成增加来纠正。产生了肌营养不良蛋白缺乏的mdx小鼠,其心肌中表达了神经元型一氧化氮合酶(nNOS)转基因。转基因的表达阻止了mdx小鼠进行性心室纤维化,并大大减少了心肌炎。通过自由活动小鼠的无线电遥测获得的心电图(ECG)显示,mdx小鼠表现出DMD患者特有的心脏异常,包括深Q波、S/R比值降低、多相R波和频繁的室性早搏。通过nNOS转基因表达,mdx小鼠的所有这些心电图异常都得到了改善或纠正。此外,nNOS转基因表达显著降低了mdx心脏自主神经功能缺陷,表现为心率变异性降低。这些发现表明,营养不良心脏增加NO生成可能具有治疗价值。
简介
心肌病经常导致杜氏肌营养不良症(DMD)患者死亡。1985年至1989年间报告的DMD死亡中,30%以上是由心力衰竭引起的(1). 随着以前致命的呼吸缺陷被呼吸机辅助设备治疗,这个数字肯定在增加(2)使心脏受累成为该病更常见的特征。目前对肌营养不良蛋白缺乏型心肌病患者的治疗选择通常仅限于使用缓解充血性心力衰竭症状的药物(三,4). 不幸的是,杜氏肌营养不良患者由于在青少年早期失去活动能力而大部分处于非活动状态,心肌病的严重程度可能会在未被注意、未经治疗的情况下发生,并导致猝死。营养不良性心肌病也影响女性营养不良蛋白突变携带者,她们通常不会出现严重的骨骼肌病变。超过50%的女性携带者经历了临床相关的心脏受累,通常严重到需要心脏移植(5,6,7).
DMD心肌病的临床和组织学表现具有特征性特征。DMD患者的心电图(ECG)显示右心前区R波高,S:R比值降低,肢体和左心前区导联Q波深,以及多相R波(8–12). 随着心肌病的进展,会出现心律失常、传导异常、局部左室壁运动异常、左室扩张和心脏自主神经功能障碍(8–10,13–20). 在6至10岁的患者中,59%的患者可通过心电图检测到临床前异常,在18岁以上的患者中100%的患者逐渐发展为临床上明显的心肌病(21). 对DMD患者心脏组织的尸检表明,严重纤维化通常局限于左心室后基底壁,是该病理学最常见的组织学特征,同时也观察到外周传导系统的纤维化(1,8–10,13,15). 此外,剩余的存活心肌细胞可能被结缔组织包裹,从而损害其细胞间连接和传导信号的能力(22). DMD患者的心脏电和功能异常被归因于心脏纤维化,这表明纤维化是DMD心肌病的重要组成部分(8).
肌营养不良蛋白缺乏mdx小鼠的心脏具有DMD心肌病的许多特征。与杜氏肌营养不良患者类似,mdx小鼠经历了心脏缺陷的渐进发展,尽管病理学不太严重。幼年成年小鼠在2-3个月龄时几乎没有病理学表现,而到12个月龄,功能和形态明显恶化(23–25). 与DMD患者一样,mdx小鼠表现出心电图异常、自主神经功能障碍、传导受损、心律失常、左心室功能恶化和扩张型心肌病(23,24,26,27). Mdx心脏也会经历严重的、进行性的结缔组织积聚(27,28),表明纤维化也可能是mdx心肌病的一些特征的原因。
病理性纤维化也是肌营养不良蛋白缺乏型骨骼肌的一个突出特征(28–30)这可能表明肌营养不良蛋白缺乏心脏和肌肉纤维化的共同机制。因为肌营养不良蛋白缺乏的骨骼肌和心脏会出现炎症(27,31–36)炎症细胞在许多病理学中促进纤维化(37–39),营养不良组织的纤维化可能在一定程度上是炎症过程的结果。这种可能性得到了以下发现的支持:mdx小鼠通过在nu/nu背景下繁殖而导致T细胞缺乏,与具有T细胞的mdx小鼠相比,显示出较少的肌肉和心脏纤维化(28). 炎症细胞可能通过分泌细胞因子(如转化生长因子-β(TGF-β))诱导纤维化,这些细胞因子刺激成纤维细胞产生结缔组织(40,41). 先前的研究人员已经表明,TGF-β在营养不良蛋白缺乏小鼠、狗和人类的骨骼肌中升高(42–44)在发生纤维化的肌肉病理阶段,TGF-β阻滞剂可以减少mdx横膈膜中胶原的表达(40).
如果心脏炎症显著导致肌营养不良蛋白缺乏心脏的心肌纤维化和随后的功能缺陷,那么影响炎症过程的干预措施在减少DMD和mdx营养不良患者的心肌病方面可能很有价值。在我们之前的研究中,我们发现肌营养不良蛋白缺乏骨骼肌的炎症因肌肉中nNOS的丢失而加剧(33),这是肌营养不良蛋白缺乏的继发后果(45,46). 在mdx小鼠骨骼肌中表达nNOS转基因以使肌肉(一氧化氮)NO生成水平正常化,从而显著降低肌肉炎症和肌膜溶解(33)这表明肌肉源性NO在营养不良骨骼肌中具有抗炎和细胞保护作用。
先前的研究人员已经表明,mdx小鼠的心脏失去了约80%的正常nNOS活性(23)类似于mdx骨骼肌中NOS活性的丧失(46). 这种相似性表明,nNOS缺陷也可能导致肌营养不良蛋白缺乏症的心肌病。然而,nNOS在心肌和骨骼肌中的分布和分子关联存在重要差异,这可能影响NO在两种肌肉类型中的功能。例如,骨骼肌中的nNOS通过与肌营养不良蛋白复合物的联系主要定位于肌膜(45). 在心肌中,nNOS位于肌膜(47)但也有报道定位于肌浆网(48)和线粒体(49,50)与肌营养不良蛋白不共存。因此,心肌中nNOS活性的缺乏可能主要反映其调节缺陷,而不是与肌营养不良蛋白复合物结合的丧失。预测心脏中NOS活性受到扰动的影响更为复杂,因为心肌中NO生成的特定部位可能对NO介导的功能产生拮抗作用。尽管内皮型NOS在肌膜产生的NO可能对心肌产生负营养不良作用,nNOS在肌浆网膜上产生的NO可对收缩力产生积极影响(51). 来自不同NOS亚型的NO-的竞争功能在骨骼肌中尚不清楚。
在本研究中,我们测试心肌细胞中nNOS对NO生成的改变是否会影响mdx心脏的病理学。在我们的假设模型中,实验性升高肌营养不良蛋白缺乏心脏的心肌NO生成将减少心脏炎症,减少纤维化,并因此使那些归因于mdx或DMD心肌纤维化的心脏功能特征正常化。我们通过生成一个心肌中表达nNOS转基因的肌营养不良蛋白缺乏小鼠系来验证我们的假设。通过对自由活动小鼠心脏功能的遥测监测,以及测试心脏炎症和纤维化的增加是否受到影响,来评估nNOS对mdx心脏中心肌NO生成增加的影响。
结果
nNOS转基因插入增加心脏组织中nNOS的表达和活性
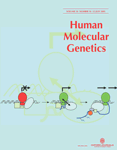
对3个月、5个月、9个月或18个月龄野生型C57小鼠和mdx小鼠的心脏进行Western blot分析表明,在所分析的任何年龄段野生型和mdx心脏的nNOS浓度没有差异(图1A显示了来自5个月大的小鼠的数据)。nNOS转基因、肌营养不良蛋白表达小鼠(nNOS Tg+小鼠)心脏中nNOS的浓度是非转基因同卵鼠(nNOS-Tg−小鼠)心脏浓度的9.4倍(图1B) 并且比nNOS转基因、肌营养不良蛋白缺乏小鼠(nNOS Tg+/mdx小鼠)的表达水平高2.0倍(图1C) ●●●●。NOS活性测定显示,相对NO产生水平通常表现出与所测定的每条线中的NOS浓度相同的趋势,尽管差异的幅度较小。活性数据是相对于nNOS Tg−心脏表达的,其平均值设定为100%(SEM=5.2;n个=9). nNOS Tg+小鼠的心脏具有51%以上的NOS活性(SEM=16.2;n个=5)比nNOS Tg−心脏和nNOS-Tg+/mdx心脏的活性高18%(SEM=2.99;n个=6)(图1D) ●●●●。然而,nNOS Tg−心脏中的NOS活性与nNOS-Tg−/mdx心脏中的活性没有显著差异(101%;SEM=3.7;n个=5).
营养不良蛋白缺乏小鼠出现心肌炎,随着NO生成的增加而减少
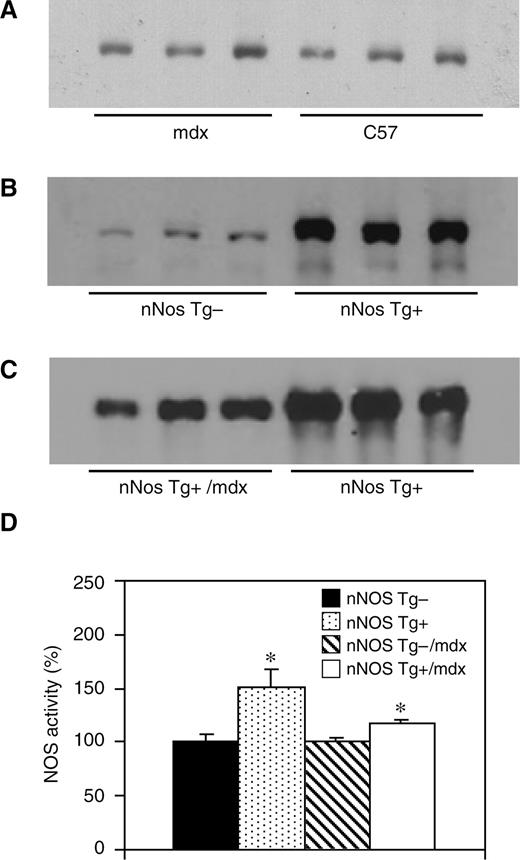
组织学分析显示,在3个月至18个月大的小鼠中,没有任何年龄段的nNOS-Tg+或nNOS-Tg−心脏发炎的证据。然而,与所有年龄段的nNOS Tg−心脏相比,nNOS-Tg−/mdx心脏含有更多的炎性细胞。在12个月龄小鼠的nNOS Tg−/mdx心脏中,巨噬细胞增加了497%(nNOS T g-/mdx=1110细胞/mm三,扫描电镜=154;nNOS Tg−=223个细胞/mm三SEM=41),嗜酸性粒细胞增加了342%(nNOS Tg−/mdx=222 单元格/mm三SEM=27;nNOS Tg−=65个电池/毫米三,SEM=14),CD4+T细胞增加了1046%(nNOS Tg−/mdx=251细胞/mm三,扫描电镜=50;nNOS Tg−=24个电池/mm三SEM=10),CD8+T细胞增加438%(nNOS Tg−/mdx=464细胞/mm三SEM=14;nNOS Tg−=106个电池/毫米三,SEM=27)(图2). 巨噬细胞是mdx心脏中主要的炎症细胞类型,通过nNOS转基因的心脏表达,其浓度显著降低。nNOS Tg+/mdx小鼠的巨噬细胞浓度降低了88%(128个细胞/mm三,SEM=26),将巨噬细胞浓度降低至与表达肌营养不良蛋白的对照组无统计学差异的水平。所分析的其他炎性细胞类型的浓度不受nNOS转基因的影响,这表明NO对巨噬细胞有特殊作用。
巨噬细胞溶解心肌细胞在体外,尽管很少有膜溶解体内
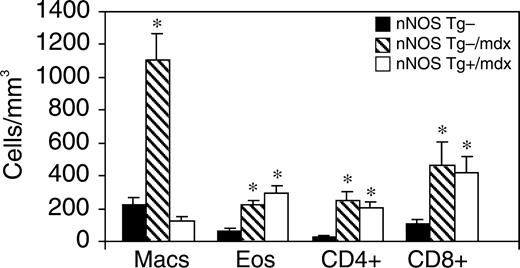
巨噬细胞介导的肌营养不良蛋白缺乏骨骼肌纤维的溶解在促进mdx骨骼肌病理学中起着重要作用(33). 我们通过使用原代心肌细胞和新分离的巨噬细胞进行共培养分析,测试心脏巨噬细胞是否也有助于mdx心肌细胞的溶解,并测试mdx心脏中的巨噬细胞浓度是否足以引起心肌细胞溶解。我们观察到,随着巨噬细胞浓度的增加,心肌细胞溶解出现剂量依赖性细胞毒性反应(图三A) ●●●●。巨噬细胞浓度与nNOS Tg−/mdx心脏中观察到的巨噬细胞浓度相当(1000个细胞/mm三),我们观察到19.4%的心肌细胞溶解,表明心肌细胞易被活化的巨噬细胞溶解。
然后,我们测试了在mdx心肌中nNOS转基因表达导致的巨噬细胞浓度降低是否会降低心肌细胞膜溶解体内试验。细胞膜受损的心肌细胞体内通过肌肉细胞质中IgG的存在进行免疫组织化学鉴定(图三B) ●●●●。在表达肌营养不良蛋白的心脏中未观察到含有IgG的肌细胞。nNOS Tg−/mdx心脏中存在含有IgG的肌细胞占据的面积,但仅占总组织面积的1.1%(SEM=0.23)(图三C) ●●●●。nNOS Tg+/mdx小鼠也可检测到含有受损心肌细胞的区域,与nNOS T g-/mdx小鼠无显著差异(占总面积的0.7%;SEM=0.18)。因此,心肌细胞膜损伤是肌营养不良蛋白缺乏心脏病理学的一个次要特征,而nNOS转基因表达或巨噬细胞浓度降低并不能显著降低心肌细胞膜损害。观察到较低水平的心肌细胞膜损伤体内与细胞毒性试验相比,mdx心脏巨噬细胞的细胞溶解性低于被激活的腹腔巨噬细胞体外试验。
表达nNOS转基因的肌营养不良蛋白缺乏心脏可预防纤维化
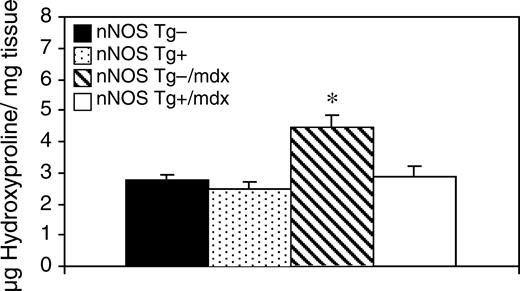
通过测量羟脯氨酸浓度来量化心脏结缔组织的总含量(52). 我们发现nNOS Tg−/mdx心脏的纤维化程度升高,其中结缔组织含量比正常对照组多60%(nNOS T g-/mdx=4.46 µg羟脯氨酸/mg组织,SEM=0.39;nNOS Tg−=2.78 µg羟脯氨酸/mg组织,SEM=0.17)(图4). nNOS Tg+/mdx小鼠的心脏纤维化程度显著低于nNOS T g-/mdx小鼠(2.86 μg羟脯氨酸/mg组织,SEM=0.33),与对照组nNOS Tg−心脏结缔组织浓度无显著差异。同样,nNOS Tg+心脏中的羟脯氨酸浓度(2.46 微克/毫克;SEM=0.27)与nNOS Tg−或nNOS-Tg+/mdx心脏中的水平无显著差异。
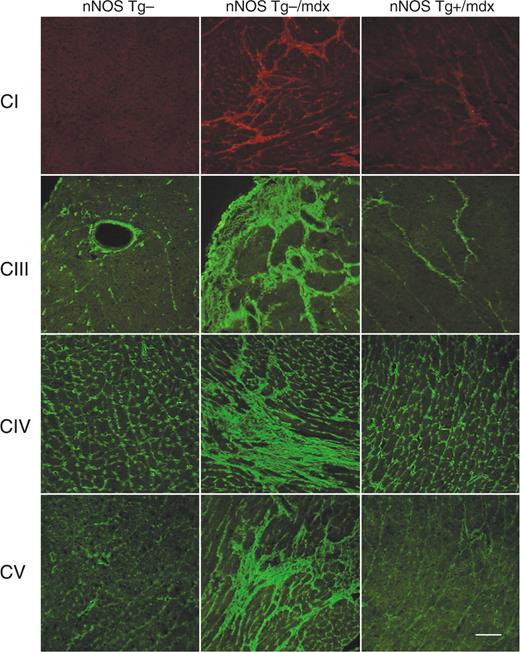
nNOS Tg−/mdx心脏组织中胶原蛋白的免疫组织学分析显示,由I、III、IV和V型胶原蛋白组成的大纤维化病变散布在心室各处(图5). 相反,nNOS-Tg+/mdx心肌中每种胶原类型的分布和浓度与nNOS-Tg−心肌非常相似,没有大的纤维化病变。nNOS Tg+小鼠的心肌未观察到纤维损伤。
nNOS转基因可减轻营养不良蛋白缺乏小鼠DMD样心电图异常和心脏自主神经功能障碍
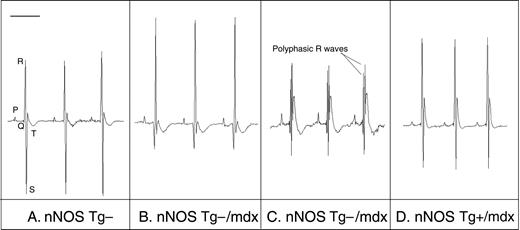
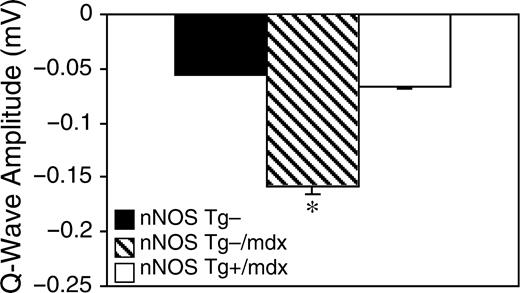
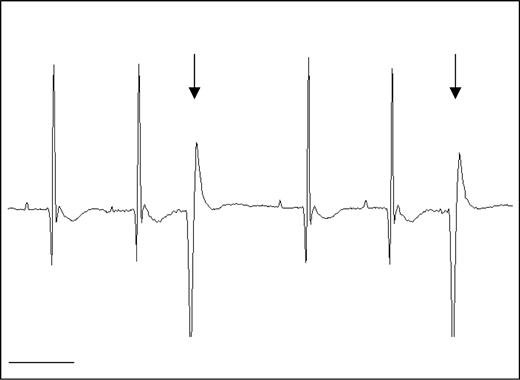
我们观察到nNOS Tg−/mdx小鼠的心电图异常,这是DMD患者的特征。nNOS Tg−/mdx小鼠的心电图描记显示深Q波(−159 µV与−61 μV(对照组)、S:R比率降低(对照组为32比54%)、多相R波和频繁心律失常(8.7比2.6/h)(图6–8和表1). 绝大多数心律失常是室性早搏(PVCs)。nNOS转基因具有显著的益处,因为nNOS Tg+/mdx小鼠表现出正常的Q波(-56 µV),S:R比率提高(38%),完全没有多相R波,心律失常频率降低到控制水平(2.7/h)。nNOS Tg−、nNOS T-g−/mdx和nNOS-Tg+/mdx小鼠之间的所有分析间隔(P–R、Q–R–S、Q–T和R–R)均相似。对nNOS Tg+小鼠的相同参数进行分析,得出的数值与nNOS T g−小鼠相当,尽管PVC型心律失常的频率远远超过nNOS t g−水平。尽管单个nNOS Tg+小鼠之间的PVC频率存在显著差异,但转基因明显具有不利影响,表明NO的过度产生可能导致心脏功能的失调。
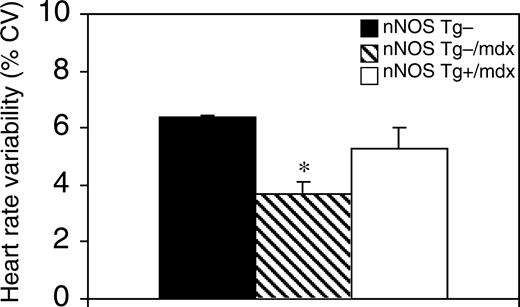
心率变异性(HRV)是心脏自主功能的指标,HRV降低表明心脏自主功能紊乱(53). 时域分析表明,nNOS Tg−/mdx小鼠的HRV降低(对照组为3.7%对6.4%,变异系数,CV),表现出自主神经异常(图9). nNOS Tg+/mdx小鼠的HRV更正常(5.3%CV),这表明nNOS转基因使心脏自主功能有些正常化,nNOS Tg−/mdx小鼠心脏中观察到的功能障碍与NO缺乏有关。
讨论
我们的数据表明,在营养不良蛋白缺乏的心脏中增加nNOS表达具有显著的有益作用。最引人注目和最有希望的发现是,nNOS转基因表达可防止营养不良蛋白缺乏心脏的纤维化。nNOS Tg+/mdx小鼠的心脏不仅在组织学上更正常,没有mdx心脏中普遍存在的大的纤维化病变,而且这些组织学改善与心电图测量的生理指标改善有关。归因于心肌纤维化的心电图异常(深Q波和S/R比降低)(8,10)完全或部分正常化。此外,在mdx小鼠中常见的多相R波(23)(本研究)在nNOS Tg+/mdx小鼠中不存在。多相R波可能表示传导异常,如传导减慢或右束支传导阻滞(10). nNOS Tg+/mdx小鼠这一特征的正常化表明,传导通路通过类似于保护心肌的机制而免受纤维化的影响,或者转基因心脏中产生的NO通过不同的机制使传导正常化。与之前公布的12个月龄mdx小鼠的心电图数据一致(23),我们没有观察到间隔时间测量的任何其他传导缺陷。我们的研究结果表明,一氧化氮介导的mdx心脏纤维化预防可导致临床上可检测到的心脏功能改善,强调了纤维化在疾病中的重要作用,并提示了新治疗策略的可能性。
nNOS Tg+/mdx心脏中心肌纤维化的减少与心肌巨噬细胞浓度的减少相对应,尽管我们不能确定mdx心肌纤维化是炎症的结果。然而,对数据的解释与许多研究的结果一致,这些研究表明NO可以抑制炎症,炎症细胞可以促进组织纤维化。NO可以减少粘附分子的表达,粘附分子介导白细胞与血管内皮的相互作用(54–56). 相反,NOS抑制增加白细胞与血管内皮细胞的粘附,从而促进炎症细胞外渗(57). 渗出后,炎症细胞可以通过多种细胞因子介导的途径中的任何一种刺激结缔组织合成,这些途径可以提高胶原蛋白和其他细胞外基质分子的表达,作为正常组织修复反应的一部分(38). 许多可由炎症细胞合成的促纤维化细胞因子在肌营养不良蛋白缺乏的肌肉中高水平表达。特别是,TGF-β参与促进mdx和DMD肌肉的纤维化(40,42–44).
nNOS转基因表达导致肌营养不良蛋白缺乏心脏中巨噬细胞浓度大幅降低,而CD8+T细胞或CD4+T细胞的浓度没有降低,这一观察结果表明,巨噬细胞在mdx心脏炎症细胞诱导的任何纤维化中起着中心作用。然而,以前的研究人员已经表明,mdx小鼠交叉进入nu/nu背景,因此它们缺乏T细胞,也显示出mdx心脏纤维化的显著降低(28). 总之,这些发现表明巨噬细胞和T细胞可能参与mdx心肌中相同的病理性纤维化途径。报告显示,从nu/nu小鼠分离的巨噬细胞对激活的反应较低,支持了这种可能性。例如,脂多糖刺激nu/nu腹腔巨噬细胞诱导的前列腺素E减少2(PGE2)野生型巨噬细胞的生成量(58). 因为PGE2可以刺激肥大细胞脱颗粒(59)肥大细胞与肌营养不良蛋白缺乏症的病理学有关(60,61),PGE水平较低2mdx-nu/nu小鼠巨噬细胞的产生可能影响肌营养不良蛋白病。另一方面,据报道,与野生型小鼠的巨噬细胞相比,nu/nu小鼠的巨噬细胞在组织中的活性更高(62–64). 因此,nu/nu小鼠缺乏T细胞会影响巨噬细胞活性的基础水平及其对进一步激活的反应,尽管尚不清楚mdx-nu/nu鼠巨噬细胞功能的T细胞调节缺失是否会增加或减少纤维化。
控制肌营养不良蛋白缺乏心脏的NO生成也可以通过与纤维化无关的过程影响心肌病。NO是心脏自主功能的重要调节器,通过对副交感神经通路的直接激动作用和对交感神经途径的间接抑制作用,共同起到防止室性心律失常的作用(65). 在NOS抑制降低副交感神经功能的动物模型中,心脏NO生成中断的有害影响显而易见(66–68),HRV降低(69)室性心律失常增加(70). 此外,心脏组织中eNOS适度过度表达增加了迷走神经驱动力,似乎具有抗心律失常作用(71). 同样,杜氏肌营养不良患者的副交感驱动力降低,交感驱动增强,心率变异性降低,窦性心动过速和室性心律失常(最显著的是室性心动过缓)频率增加,所有这些都可能反映心脏NO生成的中断(14,15,17,19,20,72,73). 我们观察到,在肌营养不良蛋白缺陷小鼠中,与DMD相关的心脏自主神经功能障碍一致的特征,如HRV降低和PVC频率增加,并发现这些异常在表达nNOS转基因的mdx小鼠中得到缓解。这些数据进一步支持了这样一种假设,即与肌营养不良蛋白缺乏相关的心脏自主神经功能障碍至少部分可归因于NO产生的中断。然而,nNOS−/-小鼠心脏出现的自主神经功能障碍(26)与mdx心脏发生的情况不同,这表明mdx心脏自主功能受损不仅仅是由于nNOS衍生NO的减少。
表达nNOS转基因的mdx小鼠中nNOS活性增加的有益效果可能高度依赖于nNOS衍生NO的生成水平。转基因在mdx心脏中的表达,其中内源性nNOS活性水平被证明降低了~80%(23),产生的总NO生成水平比健康nNOS Tg−心脏中测得的水平高18%。我们的研究结果表明,这18%的增加与心功能正常化有关。然而,我们还观察到,在表达肌营养不良蛋白的心肌中,nNOS转基因表达产生内源性nNOS衍生NO和转基因nNOS派生NO,使总NO生成增加51%。NO生成量的增加与PVC比率的大幅增加有关,因此它们的发生频率是nNOS Tg−心脏的数百倍。我们观察到nNOS Tg+心脏的心肌炎症或纤维化没有增加,这表明PVC升高是由于自主功能受损。
窦性心动过速可能由副交感神经功能受损引起,在DMD患者中常见(9,10,13,14,72,73)并且可能受到心脏中NO水平的影响。然而,大多数报告表明NO对哺乳动物心脏具有正变时作用(70,74,75)这表明nNOS活性的降低不会导致DMD心脏的心动过速。NO对心率的正性变时效应可能反映出NO对心肌细胞的直接刺激作用,而不依赖副交感系统,因为NO供体应用于切除的心房在体外可以增加自发搏动(76). 我们没有观察到mdx小鼠心率增加,尽管其他研究人员报告心率增加(23,26)或减少(24)mdx小鼠的心率。然而,由于麻醉剂的使用会影响自主功能,因此对mdx小鼠的实验治疗之间的心率数据会存在差异(77)而mdx心率反应被认为在压力条件下会受到干扰(24). 使用长期饲养在笼子中的非麻醉动物,通过遥测收集数据,提供了最能代表小鼠正常心率的数据。
尽管nNOS功能的破坏有助于肌营养不良蛋白缺乏心脏和骨骼肌的病理学,但肌营养不良素缺乏和nNOS缺陷之间的关系在骨骼肌和心脏中存在明显差异。在骨骼肌中,肌营养不良蛋白的丢失导致nNOS蛋白和mRNA的浓度大幅度降低,从而导致mdx肌肉中nNOS活性降低70%以上(46). 然而,本研究结果表明,心肌中的nNOS浓度不受肌营养不良蛋白缺乏的影响,尽管肌营养不良素缺乏的心脏中的nNO活性降低了~80%(23). 这些观察结果表明,肌营养不良蛋白缺乏导致nNOS调节缺陷,而不是心脏nNOS表达缺陷。也许,骨骼肌和心肌中肌营养不良蛋白缺乏和nNOS功能障碍之间的这些明显关系反映了nNOS和心肌中的肌营养不良素复合体之间缺乏相互作用。与骨骼肌不同,nNOS和肌营养不良蛋白复合物共同分布在表面膜上,并从中分离(45)nNOS和肌营养不良蛋白在心肌细胞中的分布并不相同。免疫组织化学观察表明,肌营养不良蛋白在T小管和心肌细胞的肌膜中富集(78),但除心肌细胞的肌膜外,nNOS还定位于肌浆网和线粒体(47–50). 此外,我们还没有发现任何已发表的结果表明nNOS与心肌中的肌营养不良蛋白复合物相关。
本研究的结果表明,正如之前在骨骼肌中观察到的那样,nNOS转基因表达在mdx心脏中的保护作用可能是通过其对巨噬细胞的抗炎作用实现的(33). 然而,我们的研究结果也表明,巨噬细胞在促进mdx骨骼肌和心肌病理方面的作用不同。例如,mdx骨骼肌纤维的溶解是肌营养不良蛋白病的一个主要特征,这主要归因于巨噬细胞的细胞溶解功能,并且由于NO生成增加而大大减少(33). 然而,在mdx心脏中,甚至在炎症区域,几乎没有证据表明心肌细胞膜溶解,NO生成增加对心肌细胞膜损伤水平没有显著影响(目前的研究)。此外,营养不良蛋白缺乏骨骼肌中NO生成的减少可以通过增加肌肉缺血来促进肌肉病理学(79,80),但mdx小鼠的心脏血管灌注正常(35,81)在DMD患者的心脏中很少有血管不规则的报道(9,10,12,15).
总之,这些发现表明,心肌nNOS活性的扰动可能有助于肌营养不良蛋白缺乏症的病理学,并表明操纵心脏NO的产生可能为减少肌营养不良蛋白缺乏症的心肌病提供一种治疗策略。然而,关于调节心脏nNOS功能和分布的微妙但重要的机制还有很多需要了解,如果生产水平大大超过正常生理生产水平,仅仅增加营养不良心肌的NO生产可能会增加心肌病。
材料和方法
动物
所有动物实验均按照美国国立卫生研究院实验动物护理和使用指南和加州大学洛杉矶分校动物护理与使用委员会的规定进行。小鼠取自我们居住在加州大学洛杉矶分校动物饲养场的繁殖群体,原始C57BL/6和从美国马萨诸塞州巴尔港的杰克逊实验室购买的mdx繁殖对。如前所述生成转基因小鼠(33)当前研究中使用的线显示nNOS转基因在心脏组织中的表达(82). 根据上述育种策略,将Mdx小鼠与nNOS Tg+小鼠杂交,产生营养不良蛋白缺乏的nNOS T g小鼠(nNOS Tg+/Mdx小鼠)(33). ARMS–PCR证实了肌营养不良蛋白的零突变(83)western blot分析证实nNOS过度表达。除非另有说明,否则使用的小鼠为混合性别,年龄为12-14个月,以确保肌营养不良蛋白缺乏品系中存在心肌病特征。
心脏nNOS表达的Western blot分析
在异氟醚诱导的安乐死后,迅速切除心脏,用pH 7.4(140)的磷酸盐缓冲盐水(PBS)冲洗 米米氯化钠,2.7 米米氯化钾,5.4 米米钠2高功率放大器4•7小时2O和1.8 米米千赫2人事军官4)并在液氮中冷冻。在40体积的十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)还原缓冲液(80 米米Tris–HCl pH 6.8,0.1 米二硫苏糖醇(DTT),70 米米SDS和1.0 米米甘油)和蛋白酶抑制剂混合物(西格玛,圣路易斯,密苏里州,美国)。样品蒸1次 min,离心1 最小值为12000克和4°C。通过在280℃下测量吸光度来测定上清液的总蛋白浓度 nm和80 根据Laemmli,在10%SDS–PAGE凝胶上分离µg每个样品(84). 蛋白质以电动方式转移到硝化纤维素膜上,同时浸入转移缓冲液(39 米米甘氨酸,48 米米Tris,0.037%十二烷基硫酸钠,20%甲醇)(85). 用0.1%Ponceau S(Sigma)染色证实了负载和转移效率的均匀性。通过在封闭缓冲液(0.5%吐温-20、0.2%明胶和3.0%干乳)中培养膜至少1次来阻断非特异性结合 h.用多克隆兔抗nNOS抗体(美国德克萨斯州德克萨斯大学健康科学中心B.S.Masters博士的慷慨捐赠)检测蛋白质样品2 h后用辣根过氧化物酶(HRP)结合的抗兔IgG(美国新泽西州皮斯卡塔韦市阿默沙姆)1 h.在培养期间,在PBS中用0.1%吐温进行一系列洗涤。通过增强化学发光(Amersham)观察条带。
NOS活性测定
通过测量[三【H】-我-精氨酸到[三【H】-我-瓜氨酸使用了Bia所述程序的改进等。(23). 冷冻的心脏在液氮下用研钵和杵碾碎,将所得粉末均匀化为5份50 米米Tris pH 7.5,含1 米米乙二胺四乙酸(EDTA),1 米米乙二醇-第二类(β-氨基醚)-N、 N,N′,N′-四乙酸(EGTA),1 米米DTT和蛋白酶抑制剂混合物(Sigma)。在1500℃离心匀浆后克和4°C,持续10 最小值,60 200μl上清液培养 µl最终浓度为50的反应缓冲液 米米Tris pH值7.5,5 米米氯化钙2, 1 米米氯化镁2, 14 µ米四氢生物蝶呤,10 µg/ml钙调蛋白,4 µ米黄素腺嘌呤二核苷酸,4 µ米黄素腺嘌呤单核苷酸,1 米米还原烟酰胺腺嘌呤二核苷酸磷酸(NADPH)和1 1微升 mCi/ml(毫升)[三【H】-我-精氨酸。30分钟后 37°C孵育min,20 米米醋酸钠pH 5.5,0.2 米米EGTA,1 米米我-瓜氨酸和2 米米EDTA并倒入Dowex-50W色谱柱(Biorad,Hercules,CA,USA),之前转化为Na+表单以分隔[三小时]-我-精氨酸[三小时]-我-瓜氨酸。洗脱液与闪烁鸡尾酒混合(美国康涅狄格州梅里登市帕卡德生物科学公司),样品在LS-3133T闪烁计数器中计数(美国加利福尼亚州富勒顿市贝克曼)。闪烁计数标准化为匀浆的总蛋白含量,通过测量280的吸光度测定 nm,表示为控制值的百分比。
组织学
在用异氟醚实施安乐死后,心脏被迅速切除,转移到充有液氮的异戊烷的小瓶中,并在−80°C下保存,直至使用。冷冻心脏通过心室短轴切成10厚的切片 微米。为了进行免疫染色,将切片风干并固定在丙酮中,用0.03%的过氧化氢熄灭内源性过氧化物酶活性。切片在含有2%明胶和3%牛血清白蛋白的PBS中培养,以防止抗体的非特异性结合。组织与一级抗体在4°C下孵育过夜,宿主适当的生物素化二级抗体(Vector Laboratories,Burlingame,CA,USA)30 室温下为min,HRP-avidin D为30 在抗体培养之间用PBS清洗切片。以3-氨基-9-乙基咔唑(AEC,红色)(载体)为底物进行染色。用于炎性细胞染色的抗体为鼠单克隆抗鼠F4/80(用于巨噬细胞)、抗鼠CD4和抗鼠CD8,均来自杂交瘤培养物的上清液(杂交瘤取自美国马里兰州贝塞斯达美国型培养物收集中心)和多克隆兔抗鼠嗜酸性粒细胞颗粒主要碱性蛋白(美国亚利桑那州斯科茨代尔梅奥诊所J.J Lee博士慷慨赠送)(86). 如前所述,通过组织形态计量学测量炎症细胞的浓度(87).
胶原染色如前所述,但有少数例外。切片与一级抗体孵育3 h,室温下,与宿主适当的荧光结合二级抗体(载体)1 h.使用的主要抗体为兔抗鼠I型胶原(Chemicon,Temecula,CA,USA)、兔抗鼠III型胶原(Chemicon)、山羊抗人IV型胶原(Southern Biotech,Birmingham,AL,USA。胶原蛋白染色切片定性分析是否存在纤维化病灶。
IgG染色
Hainsey先前通过IgG染色鉴定膜受损的心肌细胞等。(27). 心室的横截面,10 厚度为µm,培养30 在PBS中加入1%明胶溶液,以阻止非特异性结合。用PBS冲洗后,用FITC-结合小鼠抗IgG(Vector Laboratories)孵育切片1 h.进行三次PBS清洗,并在配备荧光光学的显微镜上观察和分析切片。根据之前的技术对受损细胞的面积进行量化(88); 将取样网格叠加在组织上,计算细胞IgG阳性细胞上的截获物数量,并将其表示为截获物总数的百分比。
细胞毒性分析
根据改良的Sen方案,从新生C57小鼠中进行原代心脏培养等。(89). 从2日龄C57小鼠中取出心脏,修剪结缔组织和心房。用0.2%胶原酶II(Invitrogen,Carlsbad,CA,USA)和0.6%胰酶(Sigma)在含有0.8 米米硫酸镁4,116 米米氯化钠,0.5 米米氯化钾,11 米米不2人事军官4, 5.5 米米葡萄糖和20 米米HEPES公司。细胞悬液预处理两次,每次30 每分钟为心肌细胞富集。对细胞进行计数,并将富集的心肌细胞置于含有青霉素和链霉素的4:1 DMEM/培养基199(Sigma)中,并以1800个细胞/mm的速度补充10%的FBS(Omega Scientific,Tarzana,CA,USA)2在涂有明胶的96微米板中产生了一层汇合层。培养物保持在37°C和5%CO2并在电镀后2-3天开始同步跳动。
通过改进我们先前描述的技术,获得了用于共培养的巨噬细胞(90). 在腹腔注射12%酪蛋白酸钠3天后,收集成年C57小鼠的腹腔渗出液。渗出物通过70 µm尼龙网,500粒细胞克并在0.85%NH中再次悬浮4Cl溶解红细胞。将细胞重新接种,悬浮在HBSS中,并覆盖在Histopaque 1077(Sigma)上。500离心后克用于45 min,从界面收集纯化的巨噬细胞,组织学检测其纯度>90%。
在放置心肌细胞培养物约5天后,细胞被装载51Cr(如上所述)(33)共培养16个 用0.6激活不同浓度的新鲜分离巨噬细胞 µ米项目管理局。然后对培养基进行分析51闪烁计数法释放铬。通过将0%设为51未经巨噬细胞培养的心肌细胞自发释放铬。这个51用0.1%Triton X-100(Sigma)溶解的心肌细胞将铬释放到培养基中,以确定100%的细胞毒性。
羟脯氨酸测定
根据Kivirikko技术,通过测量组织中羟脯氨酸的浓度来量化心脏结缔组织的总含量等。(52)我们以前使用过的(91).
心电图
通过植入无线电遥测设备(TA10ETA-F20,Transoma-data Science Intl.(DSI),美国明尼苏达州圣保罗),在清醒、自由移动的小鼠中收集心电图数据。测量清醒小鼠的心电图活动是有利的,因为可以消除麻醉对心脏功能的混杂影响。将传送器装置植入麻醉小鼠的腹膜腔内,并将两根电线固定在心尖和右肩峰附近,呈II导联方向。小鼠被单独安置在笼子里,通过连接到计算机系统的天线接收器进行数据记录。收集了10个未经过滤的心电图数据 s每小时35天。前7天的数据被丢弃,以便从手术过程中恢复过来,并确保麻醉效果消失。使用DSI分析软件包(ART 3.01和Physiostat 4.01)分析数据波形和参数,并对测量值进行汇编和平均,以确定心率、ECG波高和间隔时间。两名独立观察员对原始心电图波形进行心律失常扫描。
心率变异性的时域测量
CV(%)是HRV的一个指数,根据Gehrmann之前描述的连续R–R区间确定等。(53). 简言之,所有正常R–R间期的标准偏差除以所有R–R区间的平均值:SDNN/RR意思是×100.
统计
使用双尾Student’st吨-测试。这个P(P)-值设置为0.05。方差分析用于心脏遥测数据的四向分析。
致谢
作者感谢Joshua Goldhaber博士对心电图数据分析的有益讨论,以及Helen C.Chang在心电图数据分析方面的帮助。这项工作得到了美国心脏协会0325146Y(M.W.H.)、美国国立卫生研究院AR40343(J.G.T.)、AR47721(J.G.T)、肌肉营养不良协会(J.G.T.)和劳比什基金会(K.P.R.)的资助。
利益冲突声明。未申报。
图1。活性nNOS转基因在心脏组织中的表达。(A类)整个心脏匀浆显示,内源性nNOS表达在成年mdx和C57小鼠之间没有差异。左边三条车道是mdx鼠标;右三车道是C57老鼠。(B类)nNOS Tg+小鼠的整个心脏匀浆显示nNOS表达增加了9.4倍。左边三条车道是nNOS Tg−hearts;右边的三条泳道是nNOS Tg+红心。(C类). nNOS转基因在nNOS Tg+小鼠心脏中的表达是nNOS Tg+/mdx心脏中表达水平的两倍。左边的三条泳道是nNOS Tg+/mdx心脏;右侧三条车道是nNOS Tg+红心。(D类). nNOS Tg+小鼠的心脏匀浆(n个=5)总NOS活性增加51%,而nNOS Tg+/mdx心脏(n个=6)与nNOS Tg−对照心脏相比,总NOS活性增加18%(n个=9). nNOS Tg−心脏和nNOS-Tg−/mdx心脏的总NOS活性没有差异(n个=5). 星号表示与nNOS Tg−控制在P(P)<0.05. 误差条代表SEM。
图2nNOS转基因表达可降低Mdx心脏炎症。心脏组织中炎症细胞的浓度。巨噬细胞;嗜酸性粒细胞;CD4+、CD4+T细胞;CD8+、CD8+T细胞。n个=每组5只动物。星号表示在P(P)<0.05. 误差条代表SEM。
图3。mdx心肌中的巨噬细胞不能有效地进行细胞溶解。(A类)活化的巨噬细胞可以裂解培养的心肌细胞。条纹条表示nNOS Tg−/mdx心脏中的巨噬细胞浓度。结果是三个实验的平均值。误差条代表SEM(B类). 用抗鼠IgG染色的nNOS Tg−/mdx心脏切片的代表性显微照片,显示细胞膜受损的区域(括号)。棒材=70 微米。(C类). 心脏切片的IgG阳性区域表示为总切片面积。nNOS Tg−小鼠的黑条太小,无法出现在直方图中。n个=每组5只小鼠。
图4。使用nNOS转基因的mdx小鼠可以预防心脏纤维化。心脏中结缔组织的浓度。n个=每组5只小鼠。星号表示与对照组相比有显著差异,P<0.05。误差条代表SEM。
图5。显示心脏组织中结缔组织和胶原亚型分布的典型显微照片。CI,胶原蛋白1型;CIII,胶原蛋白3型;CIV胶原蛋白4型和CV,胶原蛋白5型。棒材=70 微米。
图6。来自mdx小鼠的心电图描记显示DMD样异常,其通过nNOS转基因的心脏表达而得到改善。使用植入式遥测设备收集的代表性心电描记。(A类)nNOS Tg−鼠标的正常跟踪。(B类)nNOS Tg−/mdx追踪显示深Q波、峰值R波和减弱S波。(C类)nNOS Tg−/mdx追踪显示多相R波。(D类)nNOS Tg+/mdx小鼠追踪显示标准化Q波、R波和S波。条形=100 毫秒。
图7在心脏表达nNOS转基因的mdx小鼠中,Q波振幅正常化。n个=每组3只小鼠。星号表示在P(P)<0.05. 误差条代表SEM。
图8.Mdx小鼠经常出现PVC。nNOS Tg−/mdx小鼠的代表性ECG描记显示在DMD患者中也常见的PVC(箭头)。条形=100 毫秒。
图9HRV是心脏自主功能的指标,在nNOS Tg+/mdx小鼠中正常化。n个=每组3只小鼠。星号表示在P(P)<0.05. 误差条代表SEM。
表1。NOS Tg-、NOS Tg−/mdx、NOS-Tg+/mdx和NOS Tg+小鼠的心电图参数
| . | P–R(P–R). | R–R. | 问-答. | 问-答-答. | S: R(右). | 人力资源. | 聚氯乙烯/小时. |
|---|
| nNOS Tg− | 41.7 (1.7) | 94.7(0.6) | 39.6(0.7) | 11.7(0.3) | 53.7(8.3) | 634 (3.9) | 2.7 (2.2) |
| nNOS Tg−/mdx | 43 (1) | 98.2 (2.2) | 38.7 (1.2) | 13 (0) | 31.9 (14.5) | 612 (14) | 8.7 (3.3) |
| nNOS Tg+/mdx | 40.3 (2.4) | 93.2 (2.0) | 35.7 (4.4) | 12 (0) | 38.1 (4.9) | 645 (13.8) | 2.7 (1.5) |
| nNOS时间+ | 41.7 (0.3) | 93.7 (1.0) | 38 (0.6) | 12.3 (0.3) | 42.7 (10.5) | 640 (6.4) | 1093 (1054.2) |
| . | P–R(P–R). | R–R. | 问-答. | 问-答-答. | S: R(右). | 人力资源. | 聚氯乙烯/小时. |
|---|
| nNOS Tg− | 41.7 (1.7) | 94.7 (0.6) | 39.6(0.7) | 11.7(0.3) | 53.7(8.3) | 634 (3.9) | 2.7 (2.2) |
| nNOS Tg−/mdx | 43 (1) | 98.2 (2.2) | 38.7 (1.2) | 13 (0) | 31.9 (14.5) | 612 (14) | 8.7 (3.3) |
| nNOS Tg+/mdx | 40.3 (2.4) | 93.2 (2.0) | 35.7 (4.4) | 12 (0) | 38.1 (4.9) | 645 (13.8) | 2.7 (1.5) |
| nNOS时间+ | 41.7 (0.3) | 93.7 (1.0) | 38 (0.6) | 12.3 (0.3) | 42.7 (10.5) | 640 (6.4) | 1093 (1054.2) |
表1。NOS Tg-、NOS Tg−/mdx、NOS-Tg+/mdx和NOS Tg+小鼠的心电图参数
| . | P–R(P–R). | R–R. | 问-答. | 问-答-答. | S: R(右). | 人力资源. | 聚氯乙烯/小时. |
|---|
| nNOS Tg− | 41.7(1.7) | 94.7(0.6) | 39.6 (0.7) | 11.7 (0.3) | 53.7(8.3) | 634 (3.9) | 2.7 (2.2) |
| nNOS Tg−/mdx | 43 (1) | 98.2 (2.2) | 38.7 (1.2) | 13 (0) | 31.9 (14.5) | 612 (14) | 8.7 (3.3) |
| nNOS Tg+/mdx | 40.3 (2.4) | 93.2 (2.0) | 35.7 (4.4) | 12 (0) | 38.1 (4.9) | 645 (13.8) | 2.7 (1.5) |
| nNOS时间+ | 41.7 (0.3) | 93.7 (1.0) | 38 (0.6) | 12.3 (0.3) | 42.7 (10.5) | 640 (6.4) | 1093 (1054.2) |
| . | P–R(P–R). | R–R. | 问-答. | 问-答-答. | S: R(右). | 人力资源. | 聚氯乙烯/小时. |
|---|
| nNOS Tg− | 41.7(1.7) | 94.7(0.6) | 39.6(0.7) | 11.7 (0.3) | 53.7(8.3) | 634 (3.9) | 2.7 (2.2) |
| nNOS Tg−/mdx | 43 (1) | 98.2 (2.2) | 38.7 (1.2) | 13 (0) | 31.9 (14.5) | 612 (14) | 8.7 (3.3) |
| nNOS Tg+/mdx | 40.3 (2.4) | 93.2 (2.0) | 35.7 (4.4) | 12 (0) | 38.1 (4.9) | 645 (13.8) | 2.7 (1.5) |
| nNOS时间+ | 41.7 (0.3) | 93.7 (1.0) | 38 (0.6) | 12.3 (0.3) | 42.7 (10.5) | 640 (6.4) | 1093 (1054.2) |
工具书类
1Moriuchi,T.、Kagawa,N.、Mukoyama,M.和Hizawa,K(
1993
)肌营养不良症的尸检分析。德岛医学实验杂志。
,40
,83
–93. 2Eagle,M.、Baudouin,S.V.、Chandler,C.、Giddings,D.R.、Bullock,R.和Bushby,K(
2002
)Duchenne肌营养不良患者的生存率:自1967年以来预期寿命的提高以及家庭夜间通风的影响。神经肌肉。迪索德。
,12
,926
–929. 三Ishikawa,Y.、Bach,J.R.和Minami,R(
1999
)Duchenne肌营养不良症的心脏保护。美国心脏杂志。
,137
,895
–902. 4Bushby,K.、Muntoni,F.和Bourke,J.P(
2003
)第107届ENMC国际研讨会:肌营养不良和强直性肌营养不良心脏受累的管理,2002年6月7日至9日,荷兰纳登。神经肌肉。迪索德。
,13
,166
–172. 5Hoogerwaard,E.M.,van der Woww,P.A.,Wilde,A.A.M.,Bakker,E.,Ippel,P.F.,Oosterwijk,J.C.,Majoor-Krakauer,D.F.,van-Essen,A.J.,Leschot,N.J.和de Visser,M(
1999
)Duchenne和Becker肌营养不良携带者的心脏受累。神经肌肉。迪索德。
,9
,347
–351. 6波利塔诺·L、尼格罗·V、尼格鲁·G、彼得雷塔·R、帕萨马诺·L,帕帕雷拉·S、迪·索玛·S和科米·L·I(
1996
)Duchenne和Becker肌营养不良症女性携带者心肌病的发展。日本汽车制造商协会
,275
,1335
–1338. 7Mirabella,M.,Servedei,S.,Manfredi,G.,Ricci,E.,Frustaci,A.,Bertini,E.,Rana,M.和Tonali,P(
1993
)心肌病可能是杜氏肌营养不良女性携带者的唯一临床表现。神经病学
,43
,2342
–2345. 8J.K.Perloff、W.C.Roberts、A.C.De Leon和D.O'Doherty(
1967
)Duchenne进行性肌营养不良的独特心电图。美国医学杂志。
,42
,179
–188. 9Perloff,J.K.、Henze,E.和Schelbert,H(
1984
)通过放射性核素显像研究Duchenne型肌营养不良患者局部心肌代谢、灌注和室壁运动的变化。循环
,69
,33
–42. 10斯拉克卡,C(
1968
)Duchenne进行性肌营养不良的心电图。循环
,38
,933
–940. 11G.W.Manning和G.J.Cropp(
1958
)进行性肌营养不良的心电图。英国心脏杂志。
,20
,416
–420. 12Gnecchi-Ruscone,T.、Taylor,J.、Mercuri,E.、Paternostro,G.、Pogue,R.、Bushby,K.、Sewry,C.、Muntoni,F.和Camici,P.G(
1999
)Duchenne、Becker和肌糖蛋白病患者的心肌病:冠状动脉功能障碍的作用?肌肉神经
,22
,1549
–1556. 13Perloff,J.K.、De Leon,A.C.,Jr和O’Doherty,D(
1966
)进行性肌营养不良的心肌病。循环
,33
,625
–648。 14Akita,H.、Matsuoka,S.和Kuroda,Y(
1993
)预测性心电图评分评估Duchenne肌营养不良患者的预后。德岛医学实验杂志。
,40
,55
–60. 15野村,H.和Hizawa,K(
1982
)Duchenne进行性肌营养不良症心脏传导系统的组织病理学研究。病理学报。日本。
,32
,1027
–1033. 16de Kermadec,J.-M.、Becane,H.-M.、Chenard,A.、Tertrain,F.和Weiss,Y(
1994
)Duchenne肌营养不良患者左室收缩功能障碍的患病率:超声心动图研究。美国心脏杂志。
,127
,618
–623. 17Yanagisawa,A.、Miyagawa,M.、Yotsukura,M.,Tsuya,T.、Shirato,C.、Ishihara,T.,Aoyagi,T.和Ishikawa,K(
1992
)Duchenne型肌营养不良患者心律失常的患病率及其预后意义。美国心脏杂志。
,124
,1244
–1250。 18Sanyal,S.K.和Johnson,W.W(
1982
)Duchenne进行性肌营养不良儿童的心脏传导异常:心电图特征和形态学相关性。循环
,4
,853
–863. 19Lanza,G.A.、Russo,A.D.、Giglio,V.、De Luca,L.、Messano,L.,Santini,C.、Ricci,E.、Damiani,A.、Fumagalli,G.、De-Martino,G.,Mangiola,F.和Bellocci,F(
2001
)Duchenne肌营养不良患者心脏自主功能受损:与心肌和呼吸功能的关系。美国心脏杂志。
,141
,808
–812. 20Chenard,A.A.、Becane,H.M.、Tertrain,F.、de Kermadec,J.M.和Weiss,Y.A(
1993
)Duchenne型肌营养不良症的室性心律失常:患病率、意义和预后。神经肌肉。迪索德。
,三
,201
–206. 21Nigro,G.、Comi,L.I.、Politiano,L.和Bain,R.J.I(
1990
)Duchenne型肌营养不良症心肌病的发病率和演变。国际心脏病杂志。
,26
,271
–277. 22Fenoglio,J.J.,Jr,Pham,T.D.,Harken,A.H.,Horowitz,法律公告,Josephson,M.E.和Wit,A.L(
1983
)复发性持续性室性心动过速:心动过速起源的心内膜下区域的结构和超微结构。循环
,68
,518
–533. 23Bia,B.L.,Caddisy,P.J.,Young,M.E.,Rafael,J.A.,Leighton,B.,Davies,K.E.,Radda,G.K.和Clarke,K(
1999
)杜氏肌营养不良小鼠模型中心肌nNOS降低、iNOS增加和心电图异常。分子细胞杂志。心脏病学。
,31
,1857
–1862. 24Quinlan,J.G.,Hahn,H.S.,Wong,B.L.,Lorenz,J.N.,Wenisch,A.S.和Levin,L.S(
2004
)mdx小鼠心肌病的演变:生理和形态学发现。神经肌肉。迪索德。
,14
,491
–496. 25Wilding,J.R.、Schneider,J.E.、Sang,A.E.、Davies,K.E.、Neubauer,S.和Clarke,K(
2005
)肌营养不良蛋白和MLP缺陷小鼠心脏:在形态和功能上有显著差异,但细胞骨架蛋白的积累相似。美国财务会计准则委员会J。
,19
,79
–81。 26Chu,V.、Otero,J.M.、Lopez,O.、Sullivan,M.F.、Morgan,J.P.、Amende,I.和Hampton,T.G(
2002
)mdx小鼠的心电图表现:Duchenne型肌营养不良症的心脏表型。肌肉神经
,26
,513
–519. 27T.A.Hainsey、S.Senapati、D.E.Kuhn和J.A.Rafael(
2003
)与肌营养不良相关的心肌病特征与心血管系统中缺乏肌营养不良蛋白无关。神经肌肉。迪索德。
,13
,294
–302. 28Morrison,J.、Lu,W.L.、Pastoret,C.、Partridge,T.和Bou-Gharios,G(
2000
)mdx营养不良小鼠的T细胞依赖性纤维化。实验室投资。
,80
,881
–891. 29Laws,N.和Hoey,A(
2004
)mdx小鼠脊柱后凸的进展。J.应用。生理学。
,97
,1970
–1977. 30Roig,M.、Roma,J.、Fargas,A.和Munell,F(
2004
)mdx小鼠腓肠肌群的纵向病理研究。神经病理学学报。
,107
,27
–34. 31Cai,B.,Spencer,M.J.,Nakamura,G.,Tseng-Ong,L.和Tidball,J.G(
2000
)T细胞效应器介导的穿孔素介导的细胞毒性促进肌营养不良蛋白缺乏的嗜酸性粒细胞症。美国病理学杂志。
,156
,1789
–1796. 32M.J.斯宾塞(Spencer,M.J.)、E.蒙特奇诺·罗德里格斯(Montecino-Rodriguez,E.)、K.多什金德(Dorshkind,K.)和J.G.蒂波(Tidball,J.G.)(
2001
)辅助性(CD4+)和细胞毒性(CD8+)T细胞促进肌营养不良蛋白缺乏症的病理。临床。免疫学。
,98
,235
–243. 33Wehling,M.、Spencer,M.J.和Tidball,J.G(
2001
)一氧化氮合酶转基因改善mdx小鼠的肌营养不良。J.细胞。生物。
,155
,123
–131. 34M.J.Cullen和J.J.Fulthorpe(
1975
)Duchenne型肌营养不良症的纤维分解阶段。电子显微镜研究。神经学杂志。科学。
,24
,179
–200. 35布里奇斯,L.R(
1986
)心肌坏死和炎症与mdx小鼠退行性和持续性肌病的关系。神经学杂志。科学。
,72
,147
–157. 36R.D.亚当斯(
1971
)肌肉疾病——病理学研究。
Harper and Row,Hagerstown,医学博士,第页。298
. 37Strutz,F.和Neilson,E.G(
2003
)免疫性肾损伤中纤维化机制的新见解。斯普林格·塞明。免疫病理学。
,24
,459
–476. 38Nicoletti,A.和Michel,J.B(
1999
)心脏纤维化和炎症:与血流动力学和激素因素的相互作用。心血管疾病。物件。
,41
,532
–543. 39Koehler,D.R.、Downey,G.P.、Sweezey,N.B.、Tanswell,A.K.和Hu,J(
2004
)肺炎症是囊性纤维化的治疗靶点。美国J.Respir。细胞分子生物学。
,31
,377
–381. 40Gosselin,L.E.、Williams,J.E.、Deering,M.、Braseau,D.、Koury,S.和Martinez,D.A(
2004
)营养不良肌肉中TGF-β1 mRNA表达的定位和早期过程。肌肉神经
,30
,645
–653. 41Striano,S.、Germani,A.、Di Cardok,A.、Porcelli,D.、De Mori,R.、Mangoni,A.,Napolitano,M.、Martelli,F.、Biglioli,P.和Capogrossi,M.C(
2004
)肌营养不良蛋白缺乏mdx小鼠的动脉生成和伤口修复增强。循环
,110
,3341
–3348. 42Iannaccone S.、Quattrini A.、Smirne S.、Sessa M.、de Rino F.、Ferini-Strambi L.和Nemni R(
1995
)Duchenne肌营养不良动物模型中的结缔组织增殖和生长因子。神经学杂志。科学。
,128
,36
–44. 43Bernasconi,P.、Torchiana,E.、Confalonieri,P.,Brugoni,R.、Barresi,R.,Mora,M.、Cornelio,F.、Morandi,L.和Mantegazza,R(
1995
)营养不良患者肌肉中转化生长因子-beta 1的表达与纤维化相关。纤维生成细胞因子的致病作用。临床杂志。投资。
,96
,1137
–1144. 44Passerini,L.、Bernasconi,P.、Baggi,F.、Confalonieri,P.,Cozzi,F..、Cornelio,F.和Mantegazza,R(
2002
)X连锁金毛猎犬肌营养不良症犬肌肉中的成纤维细胞因子和纤维化程度。神经肌肉。迪索德。
,12
,828
–835. 45Brenman,J.E.,Chao,D.S.,Xia,H.,Aldape,K.和Bredt,D.S(
1995
)一氧化氮合酶与肌营养不良蛋白复合,在Duchenne型肌营养不良症的骨骼肌肌膜中缺失。单元格
,82
,743
–752. 46Chang,W.-J.,Iannaccone,S.T.,Lau,K.S.,Masters,B.S.S.,McCabe,T.J.,McMillan,K.,Padre,R.C.,Spencer,M.J.,Tidball,J.G.和Stull,J.T(
1996
)神经型一氧化氮合酶与肌营养不良蛋白缺乏性肌营养不良。程序。美国国家科学院。科学。美国
,93
,9142
–9147。 47Xu,K.Y.、Kuppusamy,S.P.、Wang,J.Q.、Li,H.、Cui,H.、Dawson,T.M.、Huang,P.L.、Burnett,A.L.、Kuppusamy,P.和Becker,L.C(
2003
)一氧化氮保护心肌肌膜酶功能和离子活性转运,防止缺血诱导的失活。生物学杂志。化学。
,278
,41798
–41803. 48Xu,K.Y.、Huso,D.L.、Dawson,T.M.、Bredt,D.S.和Becker,L.C(
1999
)心肌肌浆网中的一氧化氮合酶。程序。美国国家科学院。科学。美国
,96
,657
–662. 49Kanai,A.J.、Pearce,L.L.、Clemens,P.R.、Birder,L.A.、Van Bibber,M.M.、Choi,S.Y.、de Groat,W.C.和Peterson,J(
2001
)用电化学检测鉴定分离的心肌线粒体中的神经元一氧化氮合酶。程序。美国国家科学院。科学。美国
,98
,14126
–14131. 50Elfering,S.L.、Sarkela,T.M.和Giulivi,C(
2002
)线粒体一氧化氮合酶的生物化学。生物学杂志。化学。
,277
,38079
–38086. 51巴鲁克,洛杉矶,哈里森,R.W.,斯卡夫,M.W.,罗莎斯,G.O.,卡波拉,T.P.,科贝西,Z.A.,霍拜,I.A.,莱蒙,C.A.,伯内特,A.L.,奥鲁克,B。等。(
2002
)一氧化氮通过一氧化氮合酶亚型的空间限制调节心脏。自然
,416
,337
–339. 52Kivirikko,K.I.,Laitinen,O.和Prockop,D.J(
1967
)修改尿液中羟脯氨酸的特定测定。分析。生物化学。
,19
,249
–255. 53Gehrmann,J.、Hammer,P.E.、Maguire,C.T.、Wakimoto,H.、Triedman,J.K.和Berul,C.I(
2000
)小鼠心率变异性的表型筛查。美国生理学杂志。心脏循环。生理学。
,279
,H733型
–H740。 54Kosonen,O.、Kankaanranta,H.、Uotila,J.和Moilanen,E(
2000
)一氧化氮释放化合物对人内皮细胞E-选择素表达和中性粒细胞粘附的抑制作用。欧洲药理学杂志。
,394
,149
–156. 55Liu,P.、Xu,B.、Hock,C.E.、Nagele,R.、Sun,F.F.和Wong PY(
1998
)NO调节肝脏I/R中P-选择素和ICAM-1 mRNA的表达及血流动力学改变。美国生理学杂志。
,275
,H2191型
–H2198。 56Aljada,A.、Saadeh,R.、Assian,E.、Ghanim,H.和Dandona,P(
2000
)胰岛素通过一氧化氮刺激抑制人主动脉内皮细胞细胞间粘附分子-1的表达。临床杂志。内分泌。Metab公司。
,85
,2572
–2575。 57Akimitsu,T.、Gute,D.C.和Korthuis,R.J(
1995
)骨骼肌一氧化氮生成抑制引起的白细胞粘附。J.应用。生理学。
,78
,1725
–1732. 58Vogel,S.N.、Hansen,C.T.和Rosenstreich,D.L(
1979
)先天性抗LPS、无胸腺小鼠菌株的特性。免疫学杂志。
,122
,619
–622. 59Nguyen,M.、Solle,M.,Audoly,L.P.、Tilley,S.L.、Stock,J.L.、McNeish,J.D.、Coffman,T.M.、Domrowicz,D.和Koller,B.H(
2002
)前列腺素E2介导的肥大细胞脱颗粒和IL-6生成调节所需的受体和信号机制。免疫学杂志。
,169
,4586
–4593. 60Gorospe,J.R.、Tharp,M.、Demitsu,T.和Hoffman,E.P(
1994
)肌营养不良蛋白缺乏的肌纤维容易发生肥大细胞颗粒诱导的坏死。神经肌肉。迪索德。
,4
,325
–333. 61Granchelli,J.A.、Pollina,C.和Hudecki,M.S(
1995
)表达肥大细胞活性过度的双突变mdx小鼠的Duchenne-like肌病。神经学杂志。科学。
,131
,1
–7. 62Cheers,C.和Waller,R(
1975
)先天性无胸腺“裸鼠”和致命照射小鼠中的活化巨噬细胞。免疫学杂志。
,115
,844
–847. 63Nickol,A.D.和Bonvente,P.F(
1977
)对无胸腺小鼠的细菌病原体具有异常高的天然抗性。感染。免疫。
,18
,636
–645. 64A.K.夏普和M.J.科尔斯顿(
1984
)先天性无胸腺小鼠巨噬细胞活性的调节。欧洲免疫学杂志。
,14
,102
–105. 65Chowdhary,S.和Towned,J.N(
1999
)一氧化氮在心血管控制调节中的作用。临床。科学。
,97
,5
–17. 66乔特·J.K.、丹森·E.J.F.、莫里斯·J.F.和帕特森·D.J(
2001
)神经元型NOS敲除小鼠的外周迷走神经对心率的控制受损。美国生理学杂志。心脏循环。生理学。
,281
,H2310型
–H2317。 67Mohan,R.M.、Heaton,D.A.、Danson,E.J.F.、Krishnan,S.P.R.、Cai,S.、Channon,K.M.和Paterson,D.J(
2002
)神经型一氧化氮合酶基因转移促进心脏迷走神经功能增强。循环。物件。
,91
,1089
–1091. 68Markos,F.、Snow,H.M.、Kidd,C.和Conlon,K(
2002
)一氧化氮通过麻醉犬的心脏副交感神经节的作用促进迷走神经对心率的控制。实验生理学。
,87
,49
–52. 69马科斯·F、斯诺·H·M、基德·C和康隆·K(
2001
)抑制神经性一氧化氮可降低麻醉犬的心率变异性。实验生理学。
,86
,539
–541. 70Pabla,R.和Curtis,M.J(
1995
)NO调节对大鼠离体心脏心律失常的影响。循环。物件。
,77
,984
–992. 71Massion,P.B.,Dessy,C.,Desjardins,F.,Pelat,M.,Havoux,X.,Belge,C.,Moulin,P.,Guiot,Y.,Feron,O.,Janssens,S.和Ballingand,J.L(
2004
)心肌细胞限制性内皮型一氧化氮合酶(NOS3)过度表达可减弱β肾上腺素能刺激并增强迷走神经对心脏收缩的抑制作用。循环
,110
,2666
–2672. 72Yotsukura,M.、Sasaki,K.、Kachi,E.、Sasali,A.、Ishihara,T.和Ishikawa,K(
1995
)Duchenne型进行性肌营养不良患者的昼夜节律和心率变异性。美国心脏病杂志。
,76
,947
–951. 73Yotsukura,M.、Fujii,K.、Katayama,A.、Tomono,Y.、Ando,H.、Sakata,K.,Ishihara,T.和Ishikawa,K(
1998
)Duchenne型进行性肌营养不良患者心率变异性的九年随访研究。美国心脏杂志。
,136
,289
–296. 74Kojda,G.、Kottenberg,K.和Noack,E(
1997
)一氧化氮合酶和可溶性鸟苷酸环化酶的抑制在正常大鼠心脏中诱导心脏抑制作用。欧洲药理学杂志。
,334
,181
–190. 75Musialek,P.、Paterson,D.J.和Casadei,B(
1999
)细胞外pH值的变化介导对我-精氨酸。心血管疾病。物件。
,43
,712
–720. 76Musialek,P.、Lei,M.、Brown,H.F.、Paterson,D.J.和Casadei,B(
1997
)一氧化氮可以通过刺激超极化激活的内向电流I(f)来增加心率。循环。物件。
,81
,60
–68. 77Chaves,A.A.、Dech,S.J.、Nakayama,T.、Hamlin,R.L.、Bauer,J.A.和Carnes,C.A(
2003
)年龄和麻醉对默林心电图的影响。生命科学。
,72
,2401
–2412. 78Klietsch,R.、Ervasti,J.M.、Arnold,W.、Cambell,K.P.和Jorgensen,A.O(
1993
)肌营养不良蛋白-糖蛋白复合物和层粘连蛋白共同定位于心肌的肌膜和横小管。圆形Res。
,72
,349
–360. 79Thomas,G.D.、Sander,M.、Lau,K.S.、Huang,P.L.、Stull,J.T.和Victor,R.G(
1998
)肌营养不良蛋白缺陷骨骼肌中α-肾上腺素能血管收缩的代谢调节受损。程序。美国国家科学院。科学。美国
,95
,15090
–15095。 80Sander,M.、Chavoshan,B.、Harris,S.A.、Iannaccone,S.T.、Stull,J.T.、Thomas,G.D.和Victor,R.G(
2000
)杜氏肌营养不良儿童神经型一氧化氮合酶缺陷骨骼肌的功能性肌肉缺血。程序。美国国家科学院。科学。美国
,97
,13818
–13823. 81科恩·R.D.、杜比杰·M.、摩尔·S.A.、科勒·巴斯克斯·R.、普鲁蒂·S.和坎贝尔·K.P(
2001
)在缺乏平滑肌肌多糖-肌跨复合体的小鼠模型中预防心肌病。临床杂志。投资。
,107
,1号机组
–R7。 82K.J.布伦南和E.C.哈德曼(
1993
)转基因小鼠中人α-骨骼肌肌动蛋白基因的定量分析。生物学杂志。化学。
,268
,719
–725. 83阿马尔菲塔诺(A.Amalfitano)和张伯伦(J.S.Chamberlain)(
1996
)耐mdx扩增突变系统检测,一种简单快速的基于聚合酶链反应的mdx等位基因检测。肌肉神经
,19
,1549
–1553. 84英国莱姆利(
1970
)噬菌体T4头部组装期间结构蛋白的裂解。自然(伦敦)
,227
,680
–685. 85伯内特,W.N(
1981
)“Western blotting”:蛋白质从十二烷基硫酸钠-聚丙烯酰胺凝胶电泳转移到未修饰的硝化纤维素,并用抗体和放射性蛋白A进行射线检测。分析。生物化学。
,112
,195
–203. 86Lee,J.J.,McGarry,M.P.,Farmer,S.C.,Denzler,K.L.,Larson,K.A.,Carrigan,P.E.,Brenneise,I.E.,Horton,M.A.,Haczku,A.,Gelfand,E.W.,Leikauf,G.D.和Lee,N.A(
1997
)白细胞介素-5在转基因小鼠肺上皮的表达导致哮喘的肺部病变《实验医学杂志》。
,185
,2143
–2156. 87Wehling-Henricks,M.,Lee,J.J.和Tidball,J.G(
2004
)泼尼松龙减少肌营养不良蛋白缺乏骨骼肌中炎症细胞浸润所需的细胞粘附分子。神经肌肉。迪索德。
,14
,483
–490. 88M.J.斯宾塞、M.E.玛丽诺亚和W.M.温克勒(
2000
)TNF-缺乏、肌营养不良蛋白小鼠膈肌和股四头肌的病理进展发生改变。神经肌肉。迪索德。
,10
,612
–619. 89Sen,A.,Dunnmon,P.,Henderson,S.A.,Gerard,R.D.和Chien,K.R(
1988
)SV40大T抗原表达后,终分化新生大鼠心肌细胞增殖并维持特异性分化功能。生物学杂志。化学。
,263
,19132
–19136。 90Nguyen,H.X.和Tidball,J.G(
2003
)肌肉特异性一氧化氮合酶转基因的表达可在小鼠改良肌肉使用期间预防肌肉膜损伤并减少肌肉炎症。《生理学杂志》。
,550
,347
–356. 91Koh,T.J.和Tidball,J.G(
1999
)一氧化氮合酶抑制剂减少大鼠骨骼肌中肌节的增加。《生理学杂志》。
,519
,189
–196.
©作者2005。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oupjournals.org





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}