摘要
动机
从头开始利用RNA-seq进行转录组分析为研究非模式生物的基因表达提供了一种很有前景的方法。然而,转录组组装的困难意味着组装者提供的连接常常代表转录组的断裂和不完整视图,使下游分析复杂化。我们介绍了Grouper,一种新的对来自从头开始可能属于相同转录物和基因的集合;随后可以更有力地分析这些组。当能够访问相关生物体的基因组时,Grouper可以将注释转移到从头开始组装,进一步改进了聚类。
结果
打开从头开始来自四个不同物种的组合,我们表明石斑鱼与现有的最先进方法相比,能够准确地对更多的连续图像进行聚类。这个石斑鱼pipeline能够根据contigs映射超过10%的读取,从而实现准确的下游差异表达分析。在存在密切相关的注释基因组的情况下,标记模块可以有效地将注释转移到重叠群,并利用这些信息进一步改进聚类。总体而言,石斑鱼提供完整高效的处理管道从头开始转录组集合。
1引言
测序技术的进步使得高效准确地探索遗传模型生物范围以外的转录组成为可能(Ekblom和Galindo,2011年;马里奥尼等。, 2008). 转录组测序打开了理解基因表达的大门,即使在没有高质量参考基因组的物种中,也可以理解基因表达,参考基因组构成了绝大多数已知物种(Martin和Wang,2011年). 转录组测序的一些常见用途包括变体检测、融合和选择性剪接事件发现以及差异表达分析(苏马纳等。, 2015;Stubben公司等。, 2014). 在转录序列未知的生物体中,关键的初始步骤是使用短测序读取来组装转录组。在短阅读测序中,转录组通常被测序到很深的深度,导致数千万到数亿次的阅读,然后需要组装以重建原始转录序列;一个名为从头开始组件。这些抄本稍后将作为后续分析的“参考”。例如,在差异分析管道中,首先将读取映射回组装的转录本,以推断转录本的丰度(李和杜威,2011年).
有许多流行的工具可以通过利用阅读之间的冗余和重叠来从短篇阅读中汇编全文抄本,例如Trinity(哈斯等。, 2013),绿洲(舒尔茨等。, 2012)和Trans-ABySS(罗伯逊等。, 2010). 类似地,有一些工具可以用来提高由这些方法生成的程序集的质量(卡博等。, 2017;Durai和Schulz,2016年). 然而,尽管采用了强大的算法和启发式,最终的输出序列(即contigs)通常并不代表完整的文本。换言之,许多抄本被分割成一组不重叠的连续体。这是由组装中的许多困难造成的,包括(但不限于)测序读取中的错误、由选择性剪接和同源基因引起的转录组复杂性、序列分子长度的不均匀或覆盖不足,以及用于组装的底层方法的缺陷(例如,所解决的计算问题与底层生物学不匹配的地方)。所有这些都会导致收集到输出连接体,这比实际分析的转录本集要大得多。
由于潜在的低质量从头开始组装输出,追求基因水平的差异表达分析而不是转录物水平(即重叠群水平)的分析更具前景和鲁棒性。为了获得基因水平的信息,我们需要推断出代表同一转录物的部分和代表同一基因亚型的连接,并将它们组合在一起。这就是这些方法戴维森和奥什拉克(2014),普季岑等。(2015)和斯里瓦斯塔瓦等。(2016)提议。Corset基于共有contigs计数计算层次聚类,RapClust聚类一个称为映射模糊图的稀疏图,其中节点表示共有contigs,边根据共有contig的丰富程度加权。此外,为了对这些集群和各种分析进行有意义的解释,重要的是要对从头开始程序集表示(加伯等。, 2011). 通常,我们对与所研究的非模式生物密切相关的物种的基因组或转录组进行了注释。可以利用此信息准确地注释从头开始组装并提高我们对差异表达基因功能的理解。传统上爆炸(阿尔特舒尔等。, 1990)用于执行该注释,然后完成基因本体(GO)分析(季等。, 2012;帕尔希曼等。, 2010).
目前,有几种方法可以处理从头开始组装,以改进下游分析并简化对此类分析结果的解释。然而,每种方法都有其局限性,除了紧身胸衣,没有一个工具可以为集群和注释提供完整的管道从头开始连体画。我们介绍石斑鱼它包含并改进了RapClust乐队并可用于准确高效地处理从头开始转录组组装,在几十分钟内产生成簇的连接物,并将标签从带注释的基因组转移到它们。中的底层算法石斑鱼从传输信息从头开始集合到图形上,为从头开始转录组分析超出了对组装的连续体的直接序列分析。完整的石斑鱼管道还提供可用于差异表达分析的量化信息。这个石斑鱼软件可在以下网站免费获得:https://github.com/COMBINE-lab/graper网站.
2材料和方法
2.1概述
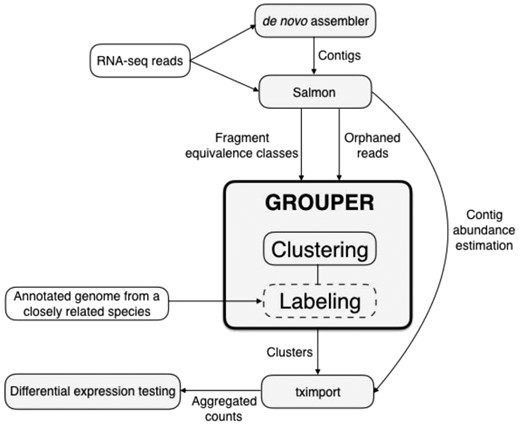
分析的第一步转录组测序在没有参考基因组的情况下,数据正在生成一个从头开始转录组汇编,使用诸如三位一体(抓斗等。, 2011)和绿洲(舒尔茨等。, 2012). 组装体往往具有大量断裂或不完整的重叠群,这些重叠群不代表全长转录物。石斑鱼目的是将这些连续体聚类,以便在理想条件下,每个聚类代表一个基因及其各种转录物。为此,原始转录组测序使用将读取映射到程序集鲑鱼生成量化估计和碎片等价类.每个等价类包含映射到来自从头开始汇编,因此编码与引用相关的排序读取的多映射结构。这些由处理石斑鱼构造映射模糊图,其中节点是重叠群的集合,并且基于它们之间的多重映射的读数通过边连接。然后使用量化估计和(可选)孤立读取信息进一步改进此图鲑鱼也可以使用石斑鱼的标记模块,将近亲物种的注释基因组添加到图表中。然后对最终的图进行聚类,提供一组连接字作为输出和注释(如果添加了)。然后可以进行下游分析,将这些群体视为假定基因。例如,使用R工具tximport(爱等。, 2017),将丰度估计值聚合到聚类水平,然后可用于差异表达测试。完整的管道如所示图1.
图1。
石斑鱼管道:石斑鱼由两个不同模块组成,群集和标记。聚类的目标是从从头开始组装成代表单个基因的组。标签模块使用密切相关物种的注释基因组为每个簇分配标签。因此,标记步骤是可选的。石斑鱼需要等价类作为输入,可以使用鲑鱼生吃的转录组测序阅读。在集群模块中可以选择使用孤立读取。的输出石斑鱼是代表假定的连续基因图谱的簇,可以使用相关物种的注释基因组进行标记。然后可以使用tximport将量化估计值求和到集群级别,以进行下游差异表达分析
中有两个主要模块石斑鱼:聚类和标记。前者将来自鲑鱼(帕特罗等。, 2017),其中转录组测序根据从头开始组装并量化。这用于生成映射模糊度图,然后对未标记的轮廓进行聚类。集群模块是石斑鱼,并且标签模块构建在该框架之上。它是可选的,并且需要从与用于装配的物种密切相关的物种中额外输入注释。这两个模块都有助于改进和注释下游分析的组装轮廓。
2.2聚类
集群模块(补充图S1)以中的工作为基础RapClust乐队,一个用于准确快速聚类来自的contigs的工具从头开始程序集(斯里瓦斯塔瓦等。, 2016). 原创作品利用了连续体内部固有的序列相似性,以等价类和转录水平的表达估计。聚类模块包括多个步骤,第一步是使用等价类,由生成鲑鱼在映射和量化阶段。图表,G公司,是一个带权的无向图,其顶点为contigs(至少映射了10个读取)。如果两个顶点共享多个映射读取,则它们由一条边连接,这意味着读取将映射到两个contigs。边缘权重表示两个contigs之间的相似程度,以及中使用的权重函数紧身胸衣(戴维森和奥什拉克,2014年),是根据这些contigs共享的读取数与分别映射到每个contigs的读取数进行定义的。一旦图形构建完成,一个额外的过滤器将通过删除连接连接contigs的边来解释来自同源基因的contigs,这些边的读取计数差异很大。或者,可以使用两个步骤进一步改进此图。第一种方法是使用孤立读取(作为输出鲑鱼),其中成对-end的末端读取映射到两个不同的contigs,具有类似的表达式估计。在第二步中,我们不是简单地删除读取计数有显著变化的相邻连续数据,而是计算一个最小割集,将包含这些连续数据的两个组件分隔开来,迫使它们在最终输出中驻留在不同的簇中。更新映射模糊度图后,使用MCL公司(Dongen,2000年)这是一种非自组织图聚类方法,用于获得表示基因连续性映射的组。
2.2.1等效等级
石斑鱼使用碎片的概念等价类估计两个碎片或连续体之间的相似性。类似的概念等价类之前已定义并用于多种用途(尼古拉等。, 2011;帕特罗等。, 2017;萨尔兹曼等。, 2011;特罗等。, 2011). 我们定义了等价类根据片段映射到的转录本,覆盖一组片段。碎片(f)我和(f)j个属于同一个等价类如果,其中是片段的成绩单集(f)我地图,以及是一组成绩单,其中(f)j个地图。每个等价类还有一个关联的计数,它只是它包含的等效片段的数量。
2.2.2图形构建
鲑鱼生成一组等价类,使用这些,我们定义了映射模糊度图,,如下所示:V(V)是一组不同的连接,C类,这是从头开始装配和边缘,,连接两个连接,,如果它们在同一个等价类.每个边缘的重量,w个伊吉,与映射到两个contigs的读取总数成比例c(c)我和c(c)j个并且与单独映射到每个contigs的读取总数成反比。我们计算w个伊吉,边缘重量e(电子)伊吉,详见方程式(1)在这个等式中,我们显示了转录本的外观c(c)我在中等价类作为一对成绩单可以出现在不同的等价类,并与多组不同的轮廓相结合,因此可以有助于多个边权重计算。使用等价类在读取映射后,我们使用等价类计算来自重叠群的读数总数的计数,因为分配给一个类别的所有读数都已映射到表示该类别的所有重叠群等价类在映射过程中。 2.2.3过滤
在构建映射模糊度图之后,我们应用了两个主要的过滤器,从紧身胸衣(Davidson和Oshlack,2014年)在第一步中,如果所有样本中映射到代表contig的读取总数小于10,则从图中删除一个节点及其所有连接。这减少了图中的噪声,因为根据映射读取的数量,这样的节点将具有高度加权的边,将其连接到相邻节点,但可能是短的或未组装的连接。第二个过滤步骤使用由鲑鱼为了减少序列相似但在不同条件下表达估计值差异显著的contigs之间的边缘数量,因此不太可能来自相同的潜在基因。该过滤器有助于去除可能来自同源基因且仅根据序列无法分离的contigs。具体而言,滤波器执行似然比测试,其中似然值按照方程式S1、S2和边的详细说明进行计算从图形中删除。
2.2.4孤儿阅读
中的第一个可选过滤器石斑鱼使用孤立读取的信息向映射模糊度图添加边。在配对测序运行中,输出是一系列读取,这样每对读取都是从来自同一转录物的单个片段中进行排序的。因此,理想情况下,这些读取应该映射到程序集中的单个contig。然而,在某些情况下,读取的一端映射到一个contig,另一端映射到不同的contig。我们称这种阅读为孤儿,因为他们的父母contig不是唯一的或可识别的。这可能是由于两个主要原因造成的。第一个是(对抗性)排序错误,这使得无法正确识别读取的原始源。第二种是装配错误或断裂的contigs,这是从头开始装配程序。由于孤立读取提供了关于哪些contigs假设来自同一转录本的额外信息,因此我们使用这些读取来连接底层映射模糊度图中的contigs,方法是在两个contigs之间添加一条边,如果它们共享一对孤立读取。只有当两个连续表达式的比率(TPM)的绝对值通过以下公式计算时,才能添加边鲑鱼,小于2。这减少了聚类方法中的假阳性数,因为孤立读取可能是由测序方法中的错误引起的,或者可能会将不同转录物或相关基因的两个连续序列错误连接在一起。以这种方式添加边有助于提高石斑鱼超过其他方法。
2.2.5 Min-cut过滤器
如前所述,第二个可选过滤器使用基于量化信息的似然比测试,但不是简单地删除轮廓之间的边缘,而是对图形执行最小割,以将两个轮廓完全分离为图形的不相交子集。这样可以确保在最后的图聚类步骤中,两个contigs不会基于连接它们的较长路径被放入同一个簇中。基于从头开始组装,这种最小切割可以提高下游差异表达测试的准确性。然而,根据底层图形结构的复杂性,这可能需要很长时间才能运行,因此是可选的。(我们注意到,我们没有尝试优化用于执行min-cut的算法,这一过程可能会大大加快。)
2.3标签
标签模块(补充图S2)被认为是一种能够通过解释从头开始程序集,而不是完整的注释管道。基于图形的方法以前也被用于传递生物数据中的信息(利布雷希特等。, 2015). 此外,该模块不仅提高了注释质量,而且还使用这些注释来改进重叠群级别的聚类,从而获得更准确的差异分析结果。我们首先标记映射模糊度图的节点,G公司这是通过将密切相关物种的注释映射到从头开始程序集,使用传统方法,如BLAST搜索(阿尔特舒尔等。, 1990). 随后,使用基于图的标签传播半监督学习方法将这些初始注释传递到图中未标记的节点。为此,我们使用了吸附算法,该算法依赖于在图形中的随机行走,并在其各种应用中用于通过各种图形有效地传播信息(巴卢哈等。, 2008). 在这个标签传播的基础上,我们构建了一个迭代算法来根据当前的标签修改图中的拓扑和边权重。重复此过程,直到图的拓扑收敛。我们方法的最终结果是为从头开始根据可用的序列和注释信息,组合和一个最好地表示这些连续体之间关系的图,然后可以像以前一样使用MCL公司.
2.3.1初始注释
初始标签可以两种格式传递到此步骤。取A作为从头开始集合,B是相关物种的注释转录本集。第一种是从a到B的简单映射。第二种格式由两个单独的文件组成,其中第一个文件包含来自核苷酸的结果爆炸的从头开始根据第二个文件中相关物种和vice-versa的注释参考构建的数据库组装测试物种。因此,第一个文件包含一个从a到B的映射,第二个文件包含从B到a的映射。在后一种情况下,这两个文件爆炸使用位分数对文件进行排序,用e值打破关系,并给contigs相应的一致性标签,概率值与序列之间的重叠长度成正比。如果没有一致意见,即如果A中的最佳命中率,则不标记contig→ B与B中最畅销的歌曲不同→ A.这个标记和映射模糊度图被传递给标记模块,然后它以迭代方式执行其算法的步骤。在算法描述中,我们使用以下符号:
是映射模糊度图迭代
E类t吨边缘设置在迭代
是来自的边E类t吨是一对未排序的
2.3.2边缘处理
标记contigs后,基于算法先前迭代的共享标签和边权重,以两种方式迭代更改图中的边:
让t吨+1表示算法的当前迭代,并让在比赛中处于领先地位E类t吨边缘。的重量e(电子),表示为,如果共享一条边的两个Contig之间存在公共标签,则可以更新,并计算为这里,对于我们的测试,α设置为0.8,是输入图形中的原始边权重(即。G公司0)和对1,对2是每个contig有标签的概率我从共享标签集合中,L(左)。我们根据测试的物种选择了α值,并表明,根据注释物种与测试物种的接近程度,该值的影响会有所不同。对于与测试物种关系较远的物种,较小的α值会产生更好的结果,因为使用序列获得的标签可能不准确,但默认情况下以1.0的概率初始化,除非用户指定其他值作为输入。
如果两个contigs很可能共享一个标签,但它们之间没有边,则会向图形中添加新边。新边权重的计算方法与上述相同。但是,代替了原始边权重(因为最初不存在边),我们使用边权重的中值将两个顶点连接到图中的相邻顶点。只有共享标签的联合概率大于0.9时,才添加边。选择此阈值是为了避免添加大量假边,尤其是在第一次迭代中,大多数标签可能被指定为1.0的概率。
2.3.3标签传播
使用来自的结果爆炸,映射模糊度图中的一部分轮廓被标记。基于图形的半监督学习算法,吸附(巴卢哈等。, 2008)[我们使用Junto库中的实现(塔卢克达尔和佩雷拉,2010年)],用于将这些标签扩展到具有大量重叠映射读取的contigs,因此在图中它们之间有一条边。该算法通过随机遍历带有标签信息的图形来工作。此信息基于与每个节点关联的三种概率进行传播:对注射,停止当前随机行走并发出新标签的概率,对abnd公司完全放弃步行的概率对续,使用当前标签继续随机行走的概率。
我们在收敛之前不会运行标签传播算法,因为我们希望在每次迭代时利用信息更新图形并更改边。因此,来自当前节点的标签仅传播到所有高度连接的邻居,从而减少了误报的数量。在标签传播结束时,与contig关联的每个标签都有一个范围内的权重,每个contig最多可以有三个标签(吸附我们采用的算法)。一旦图的拓扑收敛,我们就会像以前一样对其进行聚类。在这种情况下,我们设置的收敛标准是,当前迭代中添加的边数小于或等于上一次迭代中添加边数的5%。最后的迭代步骤还从从头开始组装到相关注释物种中的转录本或基因。因此,聚类代表了推定的重叠群到基因水平的映射,并且可以基于聚类中大多数重叠群的标签为每个聚类分配基因标签。
3结果
3.1测试设置
分析中不同模块的性能石斑鱼,我们在来自四种不同生物体的数据集上运行了该工具,这些生物体的复杂性和基因组大小各不相同。我们考虑来自人类、小鼠和酵母的数据集。为了检查装配质量的影响,我们测试了使用三位一体(抓斗等。, 2011)以及每个有机体的参考转录本(后一个实验作为我们期望该方法如何作为从头开始装配变得更加准确和完整)。我们还包括一种植物,亚洲水稻,在分析中显示石斑鱼处理包含许多具有高序列相似性的转录本的数据。在四个数据集上,我们比较了石斑鱼与contig聚类方法预测的结果相比,紧身胸衣(戴维森和奥什拉克,2014年). 所有结果包括原始结果石斑鱼算法,以及应用了可选过滤器的算法。我们将基本算法称为石斑鱼,改编自RapClust乐队(斯里瓦斯塔瓦等。, 2016)并且只使用读取计数来过滤轮廓和边,石斑鱼(O) 指的是石斑鱼考虑到孤儿阅读对,以及石斑鱼(O)+M) 指应用两个可选的后处理步骤、使用孤立读取添加边和使用读取计数对图形执行最小剪切后的结果。用于运行工具的数据集和参数的详细信息见补充材料.
3.2石斑鱼提高聚类的准确性和速度
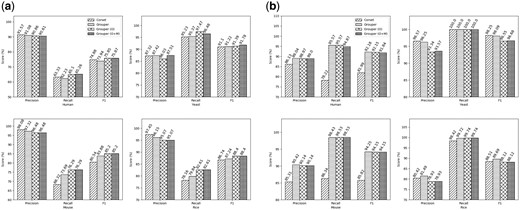
为了计算不同方法生成的簇的准确性,我们首先使用鲑鱼(帕特罗等。, 2017)(用于石斑鱼)和对齐读取使用鲍蒂(Langmead,2010年)(用于紧身胸衣). 此步骤是使用生成的两个程序集完成的三位一体和带注释的参考转录本。由于前一种情况下的“true”集群是使用爆炸(见第2节),它们不能保证没有错误。为了解释这一点,我们对参考转录组进行了相同的测试,从注释的基因组中获得了真相。这一结果表明,随着程序集质量的提高,这些聚类方法的性能可能会如何。在这两种情况下,如果同一组中的两个连续基因具有相同的“真”基因标签,则计算为真阳性,如果没有,则计算假阳性。相反,如果通过聚类方法将在真值下具有相同标签的两个重叠群放在不同的组中,则计算假阴性。
来自从头开始组件如所示图2a看F1的得分,石斑鱼表现始终优于紧身胸衣特别是包括两个额外的过滤器。一般来说,石斑鱼聚类的召回率较高,但精确度略低于紧身胸衣。虽然中的可选过滤器石斑鱼根据数据集的大小,不要对集群的准确性造成太大的差异,我们建议使用它们,因为集群中的微小差异可能导致不同的差异表达式调用。每种情况下聚集的contigs总数没有显著差异,如中的左侧面板所示补充表S1,除非是人工数据集,其中石斑鱼聚集了数量多得多的contigs。在我们的实验中石斑鱼和紧身胸衣被告知忽略映射到它们的读取数少于10的contigs(这两个工具中的默认参数)。然而,由于这两种方法使用不同的量化管道鲑鱼更高,如所示表1因此,在石斑鱼,在考虑孤立读取的计数时,图表中只包含了几个额外的连接字。
表1。上每种方法对齐的读取百分比从头开始汇编和参考转录本
| . | 从头开始程序集
. | 转录组
. |
|---|
| . | 鲍蒂. | 鲑鱼. | 鲍蒂. | 鲑鱼. |
|---|
| 人类 | 86.34 | 95.76 | 86.99 | 94.72 |
| 酵母 | 66.48 | 97.84 | 56.47 | 87.99 |
| 鼠标 | 30.74 | 86.01 | 88.2 | 85.98 |
| 大米 | 82.28 | 92.36 | 79.45 | 83.8 |
| . | 从头开始程序集
. | 转录组
. |
|---|
| . | 鲍蒂. | 鲑鱼. | 鲍蒂. | 鲑鱼. |
|---|
| 人类 | 86.34 | 95.76 | 86.99 | 94.72 |
| 酵母 | 66.48 | 97.84 | 56.47 | 87.99 |
| 鼠标 | 30.74 | 86.01 | 88.2 | 85.98 |
| 大米 | 82.28 | 92.36 | 79.45 | 83.8 |
表1。上每种方法对齐的读取百分比从头开始汇编和参考转录本
| . | 从头开始程序集
. | 转录组
. |
|---|
| . | 鲍蒂. | 鲑鱼. | 鲍蒂. | 鲑鱼. |
|---|
| 人类 | 86.34 | 95.76 | 86.99 | 94.72 |
| 酵母 | 66.48 | 97.84 | 56.47 | 87.99 |
| 鼠标 | 30.74 | 86.01 | 88.2 | 85.98 |
| 大米 | 82.28 | 92.36 | 79.45 | 83.8 |
| . | 从头开始程序集
. | 转录组
. |
|---|
| . | 鲍蒂. | 鲑鱼. | 鲍蒂. | 鲑鱼. |
|---|
| 人类 | 86.34 | 95.76 | 86.99 | 94.72 |
| 酵母 | 66.48 | 97.84 | 56.47 | 87.99 |
| 鼠标 | 30.74 | 86.01 | 88.2 | 85.98 |
| 大米 | 82.28 | 92.36 | 79.45 | 83.8 |
图2。
准确度结果:使用不同的聚类方法对从头开始程序集(一)和参考转录本(b条)来自测试物种
为了证明转录组组装方法对生成的簇准确性的影响,我们对原始转录组重复了相同的测试(结果见图2b). 就人类、老鼠和大米而言,石斑鱼性能优于紧身胸衣在精确度和召回率方面。对于酵母数据集,石斑鱼表现稍差。然而,重要的是要注意石斑鱼簇6725个连续,而紧身胸衣仅集群3592,如右面板所示补充表S2因此,石斑鱼丢弃较少的读取(表1),因此可以继续,同时保持生成簇的准确性。由石斑鱼特别是在人类和小鼠数据集的情况下,表明随着转录组组装方法的改进石斑鱼算法可能具有比现有contig聚类工具更好的性能。我们认为,这也可以减少对额外过滤器的需求。
中报告了每个聚类方法所用的时间(以秒为单位)表2和中的内存使用补充表S3。在从头开始程序集,所有变体石斑鱼比紧身胸衣,最多需要几分钟来处理输入数据并生成集群。这对于使用参考转录酶的测试也是如此,除非在人类的情况下,两个可选过滤器都已启用。此数据集上的大型运行时是由于在大型(且更密集)映射模糊度图上重复执行min-cut的计算复杂性。中的结果表2仅表示工具的集群组件所需的时间。然而,整个管道还包括根据输入引用序列对齐读取。紧身胸衣将使用Bowtie生成的对齐BAM文件作为输入,而石斑鱼将作为输入等价类由生成鲑鱼。我们报告在原始环境中运行这两个工具的时间转录组测序读入补充表S4.鲑鱼能够在几分钟内绘制并量化读数,而Bowtie可能需要几个小时。总的来说鲑鱼和石斑鱼pipeline只需几分钟就可以处理测序数据并生成集群。
从头开始装配
. | 转录组
. |
|---|
| . | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). |
|---|
| 人类 | 902.19 | 50.49 | 100.93 | 269.99 | 24479.29 | 154.46 | 296.62 | 549160.99 |
| 酵母 | 167.8 | 1.92 | 8.67 | 11.28 | 234.6 | 3.23 | 10.07 | 33.06 |
| 鼠标 | 348.91 | 14.31 | 46.12 | 46.12 | 503.54 | 27.65 | 52.65 | 52.65 |
| 大米 | 234.96 | 37.02 | 74.85 | 74.85 | 259.93 | 12.11 | 24.95 | 24.95 |
从头开始装配
. | 转录组
. |
|---|
| . | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). |
|---|
| 人类 | 902.19 | 50.49 | 100.93 | 269.99 | 24479.29 | 154.46 | 296.62 | 549160.99 |
| 酵母 | 167.8 | 1.92 | 8.67 | 11.28 | 234.6 | 3.23 | 10.07 | 33.06 |
| 鼠标 | 348.91 | 14.31 | 46.12 | 46.12 | 503.54 | 27.65 | 52.65 | 52.65 |
| 大米 | 234.96 | 37.02 | 74.85 | 74.85 | 259.93 | 12.11 | 24.95 | 24.95 |
从头开始装配
. | 转录组
. |
|---|
| . | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). |
|---|
| 人类 | 902.19 | 50.49 | 100.93 | 269.99 | 24479.29 | 154.46 | 296.62 | 549160.99 |
| 酵母 | 167.8 | 1.92 | 8.67 | 11.28 | 234.6 | 3.23 | 10.07 | 33.06 |
| 鼠标 | 348.91 | 14.31 | 46.12 | 46.12 | 503.54 | 27.65 | 52.65 | 52.65 |
| 大米 | 234.96 | 37.02 | 74.85 | 74.85 | 259.93 | 12.11 | 24.95 | 24.95 |
从头开始装配
. | 转录组
. |
|---|
| . | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). | 紧身胸衣. | 石斑鱼. | 石斑鱼(O). | 石斑鱼(运行+维护). |
|---|
| 人类 | 902.19 | 50.49 | 100.93 | 269.99 | 24479.29 | 154.46 | 296.62 | 549160.99 |
| 酵母 | 167.8 | 1.92 | 8.67 | 11.28 | 234.6 | 3.23 | 10.07 | 33.06 |
| 鼠标 | 348.91 | 14.31 | 46.12 | 46.12 | 503.54 | 27.65 | 52.65 | 52.65 |
| 大米 | 234.96 | 37.02 | 74.85 | 74.85 | 259.93 | 12.11 | 24.95 | 24.95 |
3.3检测差异表达基因的准确性
转录组组装方法往往会产生不完整或断裂的contigs,最终混淆下游差异表达测试。因此,重要的是,通过处理组装的连接体产生的簇接近实际的基因级表达估计值。为了测试这一点,我们对不同方法生成的簇进行差异表达分析。每个簇都有一个基因标签,这是通过进行连续基因映射并用其组成的连续基因中最常见的基因标签标记簇而获得的。集群级表达式估计紧身胸衣作为工具本身的输出提供石斑鱼,使用R包tximport获得(爱等。, 2017),将提供的读取计数相加鲑鱼到集群级别。然后,Limma Voom公司(法学等。, 2014)用于获得更正P(P)-每个簇是差异表达的假设值,用校正后的基因调用P(P)-差值小于或等于0.05。相同的过程,使用来自鲑鱼或RSEM(RSEM)(李和杜威,2011年)重复“真”聚类以获得基本事实。
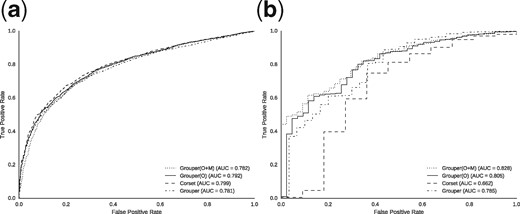
我们在人类和酵母数据集上重复了这个测试转录组测序不同条件下的样品。请注意,其他数据集仅包含来自单个条件的样本,因此不能用于差异表达分析。该分析的结果见图3中的、和补充图S3曲线代表了不同方法的准确性。在人类数据集中石斑鱼执行类似于紧身胸衣尽管其AUC略低。另一方面在酵母数据集中,石斑鱼变体的性能比紧身胸衣,与两个可选过滤器一起运行时,性能几乎提高了1.25倍。这表明石斑鱼始终如一地生成良好的聚类,不仅在实际聚类的准确性方面,而且在对其执行的下游分析方面。有趣的是,这项测试还表明,聚类本身的准确性并没有立即或成比例地反映在差异基因表达测试的准确性中(至少在这里使用的测试方案下)。
图3。
DGE结果:曲线表示使用鲑鱼计算为基本事实,由人类中每种方法生成的簇表示(一)和酵母(b条)数据集
3.4合并注释成绩单中的信息
为了对差异表达分析进行有意义的解释,需要有一些概念,即哪些基因是由单个连续基因表示的。通常,我们已经注释了与组装的非模型生物体密切相关的物种的基因组。可以收集来自该注释的信息,以准确地注释来自从头开始组件。传统上爆炸用于传输这些注释。紧身胸衣还提供了一种方法,通过将读取内容与带注释的转录组对齐并一起处理所有对齐文件来传输注释。我们在中提供标签模块石斑鱼它允许高效地将注释传输到从头开始组装,然后通过映射模糊图传播它们,以最终注释更多的连续图。
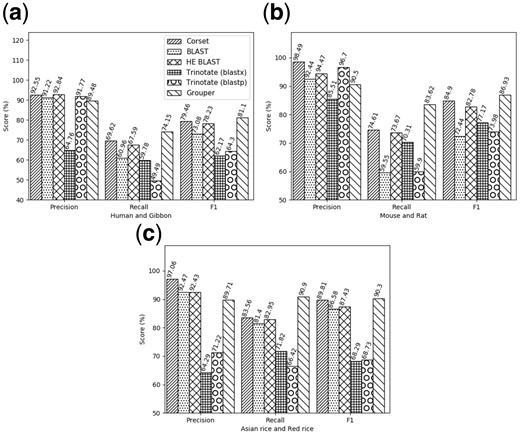
我们将结果与紧身胸衣,爆炸使用组件中的所有轮廓,爆炸仅使用具有大于10个读取映射的contigs,以及特里诺塔特(哈斯等。, 2013),同时使用核苷酸和蛋白质水平爆炸针对SwissProt数据库。由于在相关物种的contigs和注释之间没有明显和完整的映射,因此我们选择比较不同的方法,根据它们为单个基因的Contig提供相同标签的能力,然后将它们聚集在一起进行下游分析。我们使用人类、小鼠和亚洲水稻数据集的注释的、密切相关的基因组进行了分析。使用的注释基因组分别来自长臂猿、鼠类和红米。结果显示于图4,显示尽管标签模块生成的簇的精度石斑鱼略低于排名第二的方法,回忆的收益显著高于排名第二,导致F1总得分较高的聚类。
图4。
准确性结果:使用长臂猿作为密切相关的注释物种,对人类基因组进行不同注释方法的精确度、召回率和F1得分(一),在使用大鼠基因组的小鼠数据集上(b条),以及使用红米基因组的亚洲水稻数据集(c(c)). 注:HE BLAST指的是使用BLAST仅注释具有10个以上读数的重叠群,通过鲑鱼
除了准确之外,石斑鱼将注释分配给更多的连续对象,而不是简单地使用爆炸这是标记传播步骤的一个优点,该步骤在每次迭代中都会利用前面的信息继续标记图中的连接组件。这意味着,一些之前未注释的连续体,无法仅根据序列相似性进行注释,将在迭代过程结束时进行标记。这样,石斑鱼能够分别为人类、小鼠和水稻组合中的2325个、4957个和3655个额外重叠群分配注释。更多注释可能有助于特定基因的下游分析。另一方面,紧身胸衣不会直接标记从头开始程序集,但将带注释的抄本与它们一起添加到集群中。然后可以使用这些集群级信息推断注释,这增加了管道的复杂性。
在生成更完整、更准确的注释的同时,石斑鱼也比其他工具快得多,如所示表3.所用时间石斑鱼包括运行时间鲑鱼(生成连续水平丰度估计值),以生成等价类,构建映射模糊度图,运行爆炸要生成初始contig标签,然后传播标签,请更改图形拓扑并对contig进行聚类。同样,紧身胸衣计时包括将每个样本中的读数与参考值对齐的时间。石斑鱼需要20 人类中的最小值,共有2320万次读取6个样本和107 389个contigs从头开始组件。其他物种需要少于15 min,小鼠共有1050万次阅读,75727次接触,大米共有800万次阅读和99745次接触。相比之下,其他工具在这些数据集上运行需要几个小时。
表3。用于在中注释contigs的每个管道的总时间(分钟)从头开始装配
| . | 紧身胸衣. | 三位一体(核). | Trinotate(保护). | 石斑鱼. |
|---|
| 人类 | 513.28 | 2356.13 | 781.56 | 20.85 |
| 鼠标 | 272.88 | 1616.01 | 498.3 | 10.23 |
| 大米 | 208.79 | 983.37 | 424.02 | 11.19 |
| . | 紧身胸衣. | 三钠(核素). | Trinotate(保护). | 石斑鱼. |
|---|
| 人类 | 513.28 | 2356.13 | 781.56 | 20.85 |
| 鼠标 | 272.88 | 1616.01 | 498.3 | 10.23 |
| 大米 | 208.79 | 983.37 | 424.02 | 11.19 |
表3。用于在中注释contigs的每个管道的总时间(分钟)从头开始装配
| . | 紧身胸衣. | 三位一体(核). | Trinotate(保护). | 石斑鱼. |
|---|
| 人类 | 513.28 | 2356.13 | 781.56 | 20.85 |
| 鼠标 | 272.88 | 1616.01 | 498.3 | 10.23 |
| 大米 | 208.79 | 983.37 | 424.02 | 11.19 |
| . | 紧身胸衣. | 三位一体(核). | Trinotate(保护). | 石斑鱼. |
|---|
| 人类 | 513.28 | 2356.13 | 781.56 | 20.85 |
| 鼠标 | 272.88 | 1616.01 | 498.3 | 10.23 |
| 大米 | 208.79 | 983.37 | 424.02 | 11.19 |
4讨论
转录组分析转录组测序仍然是一个挑战,尤其是当被检测的生物体缺乏高质量的参考转录组时。在可用的测序技术(提供更高深度和更高质量的读取信息)和用于根据测序实验的输出重建潜在转录组的计算方法方面,已经取得了相当大的进展。然而,即使是最好的组装方法也常常会产生不完整(有时甚至不正确)的转录序列。这些组装体的断裂性质使下游分析(如差异表达分析)复杂化,并导致虚假的假阳性呼叫,以及在可以真正确定为差异表达的基因方面的统计能力损失。
遵循最初由制定的总体框架戴维森和奥什拉克(2014),我们介绍石斑鱼,用于处理的工具从头开始根据共享序列的证据,将连续体组合成假定基因的转录组数据。这些假定的基因可以更准确、更稳定地量化。我们将确定转录组的问题归结为一个聚类映射歧义图的问题,该图是使用多映射读取信息构建的。我们引入了有用的启发式方法,用于过滤此图(以删除可能的伪边),并使用从孤立映射中获得的信息来增强此图,这些孤立映射可能是由基础程序集的不完整性造成的。我们证明了这个聚类问题可以有效而准确地解决,并且将得到的聚类作为假定的基因处理可以导致有生物学意义的分析。
同时,我们还介绍了一种新颖的基于图形的方法,用于在从头开始利用相关生物的信息进行组装。我们不仅依赖于两个参考文献之间的序列相似性,还利用了映射模糊度图,该图提供了连续序列相似性的证据在内部组件。我们首先用从相关生物体转移来的高置信度标签来标记连续体的子集(图中的节点)。然后,我们通过应用半监督标签传播算法来利用图结构(塔卢克达尔和佩雷拉,2010年),在集合内高度相关的contigs之间传播基因标签。我们证明,当包含来自相关生物的标记信息时,基于基于图形的标记传播方法的标记方案适度优于其他方法,以结果簇的质量衡量(图4). 因此,我们表明石斑鱼为提高从头开始转录组分析,通过允许连接物聚集成具有生物学意义的组(假定基因),并且在可用时可以有效利用相关转录组。鉴于其相对温和的计算开销,我们认为石斑鱼可以成为一种流行的工具,帮助解决从头开始转录组分析。
While期间石斑鱼在我们的测试中表现良好,能够对来自从头开始程序集的效率很高,基于底层的质量和完整性,它所能获得的准确性显然是有限的从头开始组件。这在中进行了演示图2,我们看到石斑鱼(和紧身胸衣)当它们被提供有参考组件作为输入时。分析结果表明,提高性能的一个主要限制因素实际上是从头开始正在生产的程序集。While期间从头开始众所周知,转录组组装是一个非常具有挑战性的问题,但无论是在正在开发的计算方法方面,还是在涉及该问题的生物技术方面,都取得了进展。未来,我们有兴趣将石斑鱼可以通过长读转录组测序(例如通过PacBio Iso-Seq或nanopore的直接RNA测序技术)提供的证据。虽然这些技术通常无法获得与传统RNA-seq相同的覆盖深度,因此不太可能测序稀有亚型,但它们提供了有关表达转录物的高质量结构信息,我们预计,这些数据将能够提供有关“主干”基因结构的信息,通过该结构可以对较短的连续基因进行分组。随着组件质量的提高,我们还建议使用石斑鱼没有包含孤立读取信息的附加过滤器。
基金
这项工作得到了美国国家科学基金会生物基础设施部的支持(奖项编号:1564917)。
利益冲突:未声明。
工具书类
阿尔特舒尔
S.F.公司。
等(
1990
)基本本地对齐搜索工具
.分子生物学杂志
。,215
,403
–410
.
巴卢哈
美国。
等(
2008
). YouTube的视频建议和发现:在视图图中随机漫步。在:第十七届万维网国际会议记录第895-904页。ACM,中国北京。
卡博
C、。
等(
2017
)压缩和纠正三位一体和绿洲rna-seq从头组装
.同行J
,5
,e2988。
戴维森
N.M.(最小值)。
,奥什拉克
答:。
(
2014
)胸衣:实现从头组装转录组的差异基因表达分析
.基因组生物学
。,15
,410
东恩
S.V.公司。
(
2000
). 通过流模拟进行图形聚类。博士论文,标准化和知识转移。
杜赖
D.A.公司。
,舒尔茨
M.H.医学博士。
(
2016
)从头转录组组装的知情k mer选择
.生物信息学
,32
,1670
–1677
.
埃克布洛姆
对。
,加林多
J。
(
2011
)下一代测序在非模式生物分子生态学中的应用
.遗传
,107
,1
加伯
M。
等(
2011
)使用rna-seq进行转录组注释和定量的计算方法
.自然方法
,8
,469
–477
.
抓斗
M.G.公司。
等(
2011
)无参考基因组的rna-seq数据的全长转录组组装
.自然生物技术
。,29
,644
–652
.
哈斯
B.J.公司。
等人(
2013
)基于rna-seq的从头转录序列重建:参考生成和三位一体分析
.自然协议
。,8
,1494
.
季
第页。
等(
2012
)鲤鱼转录组特征:测序、从头组装、注释和比较基因组学
.公共科学图书馆一号
,7
,e35152。
朗米德
B。
(
2010
)将短测序读数与领结对齐
.货币。协议。生物信息学
第11章第11.7单元。
法学
C.W.公司。
等(
2014
)Voom:精确权重解锁用于rna-seq读取计数的线性模型分析工具
.基因组生物学
。,15
,R29。
锂
B。
,杜威
C.N.公司。
(
2011
)Rsem:有或无参考基因组的rna-seq数据的准确转录定量
.BMC生物信息
。,12
,323
利布雷希特
M.W.公司。
等(
2015
)染色质状态和染色质构象的联合注释揭示了结构域类型之间的关系,并确定了细胞类型特异性表达的结构域
.基因组研究
。,25
,544
–557
.
马里奥尼
J.C.公司。
等(
2008
)Rna-seq:技术再现性评估和与基因表达阵列的比较
.基因组研究
。,18
,1509
–1517
.
马丁
联合国。
,王
Z.公司。
(
2011
)下一代转录组组装
.自然版本基因
。,12
,671
–682
.
尼古拉
M。
等(
2011
)从rna-seq数据估计选择性剪接亚型频率
.算法。分子生物学
。,6
,9
帕尔希曼
T.L.公司。
等(
2010
)生态重要树种的转录组测序:组装、注释和标记发现
.BMC基因组学
,11
,180
帕特罗
对。
等(
2017
)鲑鱼提供快速且有偏差的转录表达量化
.自然方法
,14
,417
–419
.
普季岑
答:。
等(
2015
)没有参考基因组的rnaseq转录物草案的语义组装和注释
.公共科学图书馆一号
,10
,e0138006。
罗伯逊
G.公司。
等(
2010
)rna-seq数据的从头组装和分析
.自然方法
,7
,909
–912
.
萨尔兹曼
J。
等(
2011
)rna-seq数据的统计建模
.统计科学
。,26
,62
舒尔茨
M.H.医学博士。
等人(
2012
)绿洲:跨越表达水平动态范围的稳健从头开始rna-seq组装
.生物信息学
,28
,1086
–1092
.
索内松
C、。
等(
2015
)RNA-seq差异分析:转录水平估计改善基因水平推断
.F1000分辨率
,4
.
苏马纳
国际卫生组织。
等(
2015
)Rna-seq从头组装揭示了感染布氏冈比亚锥虫的冈比亚舌鳎与未感染和自愈苍蝇的差异基因表达
.前面。微生物
。,6
.
斯里瓦斯塔瓦
答:。
等(
2016
). 使用片段等价类对从头转录组进行准确、快速和轻量级的聚类。arXiv预打印arXiv:1604.03250。
Stubben公司
C.J.公司。
等(
2014
)泰国伯克霍尔德菌小rna在不同环境和胁迫条件下的差异表达
.BMC基因组学
,15
,385
塔卢克达尔
P.P.公司。
,佩雷拉
F、。
(
2010
). 用于类实例获取的基于图形的半监督学习方法的实验。在:计算语言学协会第48届年会会议记录第1473-1481页。计算语言学协会,瑞典乌普萨拉。
特罗
E。
等(
2011
)使用多映射rna-seq读取估计单倍型和亚型特异性表达
.基因组生物学
。,12
,R13。
©作者2018。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oup.com








{kind=link}
{kind=link}
{kind=link}
{kind=link}