摘要
动机
为了深入了解蛋白质在细胞中的功能,了解其亚细胞定位是必不可少的。目前的研究主要集中在仅基于序列信息的人类蛋白质亚细胞位置预测。尽管在这方面已经作出了相当大的努力,但这个问题远未得到解决。大多数现有方法只能用于处理单位置蛋白质。实际上,具有多个位置的蛋白质可能具有一些特殊的生物功能,这些功能对基础研究和药物设计都特别重要。
结果
利用多标记理论,我们通过将关键的GO(基因本体)信息提取到一般PseAAC(伪氨基酸组成)中,提出了一种新的预测因子“pLoc-mHum”。在同样严格的基准数据集上进行的严格交叉验证表明,所提出的pLoc-mHum预测因子在预测人类蛋白质亚细胞定位方面明显优于iLoc-Hum这一最先进的方法。
1引言
细胞和分子生物学的基本问题之一是理解细胞作为生命基本单位的工作过程。要真正理解这一点,必须了解不同细胞器(或亚细胞位置)中的蛋白质。
在过去二十年左右的时间里,开发了许多计算方法来解决这个问题(参见(蔡和周,2000;塞达诺等。, 1997;周和蔡,2002;周和埃尔罗德,1998年,1999年a,b条;丁和张,2008;埃马努埃尔松等。, 2000;Gardy,2003年;Nanni和Lumini,2008年;莱因哈特和哈伯德,1998年)以及两篇评论文章(周和沈,2007c;Nakai,2000年)以及其中引用的一长串参考文献]。
但上述所有方法都是在假设细胞中的每个蛋白质都有且只有一个位置的情况下发展起来的。换句话说,在这些方法中,该主题被简化为一个单标签系统。然而,随着更多实验数据的出现,蛋白质在细胞中的定位实际上是一个多标记系统,其中一些蛋白质可能同时出现在两个或多个不同的定位位点中(或在它们之间移动)。这种复合蛋白质通常具有一些特殊的生物学功能(Glory and Murphy,2007年;Shen和Chou,2007年)不应忽视,因为它们对于深入了解细胞内的生物过程和开发多靶点药物非常重要。
大约10 多年前,人们已经做出了相当大的努力来探索这种多重蛋白质系统(周和申,2007a,2010年a,b条;周等。, 2011,2012;黄和袁,2013;林等。, 2013;梅,2012;Pacharawongsakda和Theeramunkong,2013年;沈和周,2009,2010年a,b条;吴等。, 2011,2012;肖等。,2011年a). 它们大致可以分为两个系列(周,2015):“PLoc”系列和“iLoc”系列。
与单标签系统相比,处理多标签系统将更加困难和复杂。特别是,对于多标签预测器来说,很难产生“绝对真实”率的下降结果。原因如下。假设一个人类蛋白质被标记为“细胞质和细胞核”,这意味着它可能同时位于现实世界中的两个细胞器或亚细胞位置。如果预测结果为“细胞质”或“细胞核”或“细胞质、细胞核和突触”,则绝对真实率不加任何分数。当且仅当预测结果也是“细胞质和细胞核”,即与实际标签完全相同时,将加1分以推导绝对真实率。因此,它是衡量多标签预测器质量的最严格的指标(周,2013). 这就是为什么在提出多标签预测因子时,许多作者(黄和袁,2013;Pacharawongsakda和Theeramunkong,2013年;王等。, 2013)甚至选择不提及“绝对真实率”这一术语。
在这项研究中,我们使用了多标签理论(周,2013)开发一种新的预测器来识别人类蛋白质的亚细胞定位,以提高其绝对正确率和绝对错误率,这是多标记预测器最重要和最苛刻的两个指标(周,2013).
2材料和方法
2.1基准数据集
根据周的五步规则(周,2011)对于开发统计预测器,首要的是构造或选择有效的基准数据集来训练和测试模型。在文献中,基准数据集通常由训练数据集和测试数据集组成:前者用于训练建议的模型,而后者用于测试模型周和沈(2007c),如果通过折刀或二次抽样(K-fold交叉验证)测试对模型进行测试,那么使用一个高质量的基准数据集就足够了,因为这样得到的结果实际上是来自许多不同独立数据集测试的组合。在本研究中,基准数据集取自(周等。, 2012;沈和周,2009). 这样做的原因如下。(i) 该数据集包含大量经实验证实的人类蛋白质,包括单位置蛋白质和多位置蛋白质,其中没有一种蛋白质具有统计学意义 将序列标识与同一子集中的任何其他序列标识配对。(ii)它也是用于训练和测试iLoc-Hum的相同基准数据集(周等。, 2012)是该领域最先进的预测工具,因此将有助于在相同的基础上进行比较。为了方便读者,中给出了基准数据集补充材料S1。它包含根据亚细胞位置将不同的人类蛋白质序列分为14个亚群。这些蛋白质在14个亚细胞位置的整体视图见补充材料S2从中我们可以看出,在3106个不同的蛋白质中,2580个属于一个位置;480至两个位置;43至三个位置,三至四个位置,无至五个或更多位置。
A breakdown of the
N个(seq)=3106人类蛋白质,根据它们在14种不同亚细胞定位中的出现情况,见表1,其中是“虚拟蛋白质”的总数(沈和周,2009)或“定位蛋白”(周等。, 2012)在基准数据集中,以及是标记在k个-对不同的蛋白质样本进行测序。因此,多重度MD(林等。, 2013)当前基准数据集的| 子集. | 亚细胞位置名称. | 蛋白质数量. |
|---|
| 中心体 | 77 |
| 细胞质 | 817 |
| 细胞骨架 | 79 |
| 内质网 | 229 |
| 内吞体 | 24 |
| 细胞外 | 385 |
| 高尔基器械 | 161 |
| 溶酶体 | 77 |
| 微粒体 | 24 |
| 线粒体 | 364 |
| 核心 | 1021 |
| 过氧化物酶体 | 47 |
| 质膜 | 354 |
| 突触 | 22 |
| 虚拟蛋白质总数N个(维拉)一 | 3681 |
| 不同序列的蛋白质总数N个(顺序) | 3106 |
| 多重度MDb条 | 1.185b条 |
| 子集. | 亚细胞位置名称. | 蛋白质数量. |
|---|
| 中心体 | 77 |
| 细胞质 | 817 |
| 细胞骨架 | 79 |
| 内质网 | 229 |
| 内吞体 | 24 |
| 细胞外 | 385 |
| 高尔基器械 | 161 |
| 溶酶体 | 77 |
| 微粒体 | 24 |
| 线粒体 | 364 |
| 核心 | 1021 |
| 过氧化物酶体 | 47 |
| 质膜 | 354 |
| 突触 | 22 |
| 虚拟蛋白质总数N个(维拉)一 | 3681 |
| 不同序列的蛋白质总数N个(顺序) | 3106 |
| 多重度MDb条 | 1.185b条 |
| 子集. | 亚细胞位置名称. | 蛋白质数量. |
|---|
| 中心体 | 77 |
| 细胞质 | 817 |
| 细胞骨架 | 79 |
| 内质网 | 229 |
| 内吞体 | 24 |
| 细胞外 | 385 |
| 高尔基器械 | 161 |
| 溶酶体 | 77 |
| 微粒体 | 24 |
| 线粒体 | 364 |
| 核心 | 1021 |
| 过氧化物酶体 | 47 |
| 质膜 | 354 |
| 突触 | 22 |
| 虚拟蛋白质总数N个(维拉)一 | 3681 |
| 不同序列的蛋白质总数N个(顺序) | 3106 |
| 多重度MDb条 | 1.185b条 |
| 子集. | 亚细胞位置名称. | 蛋白质数量. |
|---|
| 中心体 | 77 |
| 细胞质 | 817 |
| 细胞骨架 | 79 |
| 内质网 | 229 |
| 内吞体 | 24 |
| 细胞外 | 385 |
| 高尔基器械 | 161 |
| 溶酶体 | 77 |
| 微粒体 | 24 |
| 线粒体 | 364 |
| 核心 | 1021 |
| 过氧化物酶体 | 47 |
| 质膜 | 354 |
| 突触 | 22 |
| 虚拟蛋白质总数N个(维拉)一 | 3681 |
| 不同序列的蛋白质总数N个(顺序) | 3106 |
| 多重度MDb条 | 1.185b条 |
为了简化后面的描述,基准数据集表示为,可以进一步表示为哪里仅包含来自“中心体”位置的人类蛋白质样本(参见。表1),仅包含来自“细胞质”位置的内容,依此类推;表示集合论中“并集”的符号。 2.2蛋白质样品配方
现在让我们考虑五步规则的第二步(周,2011); 即如何用一个有效的数学表达式来表示生物序列样本,该表达式能够真实地反映其与相关目标的本质相关性。给定人类蛋白质序列P(P),其最直接的表达式是哪里L(左)表示蛋白质的长度或其组成氨基酸残基的数量,是第一个残留物,第二个残渣,第三个残留物等等。由于所有现有的机器学习算法,如SVM(支持向量机)(陈等。,2016年a),KNN(K-最近邻)(肖等。, 2013),RF(随机森林)(贾等。,2016年c)和PCA(主成分分析仪)(杜等。, 2017)只能处理向量(周,2015),我们必须转换公式4变成一个向量。但在离散模型中定义的向量可能会完全丢失所有序列序信息。为了解决这个问题,PseAAC(Pse(磅/平方英寸)乌多A类米诺A类cid公司C类复合材料)(周,2000,2001,2005). 自从周氏PseAAC提出伪氨基酸组成概念以来(曹等。, 2013;杜等。, 2012;Lin和Lapointe,2013年)它被提出后,已被广泛应用于许多生物医学和药物开发领域(钟和周,2014;周和钟,2016)以及计算蛋白质组学的几乎所有领域(艾哈迈德等。, 2015;埃斯马埃利等。, 2010;贾等。,2016年b;梅赫尔等。2017年;穆罕默德等。, 2011;穆罕默德·贝吉等。, 2011;Mondal和Pai,2014年;Nanni和Lumini,2008年;南尼等。, 2012;Pacharawongsakda和Theeramunkong,2013年;拉希米等。, 2017;2010年《莎湖与熊猫》;特里帕西和潘迪,2017年;周等。2007年)以及两篇综述论文中引用的一长串参考文献(周,2009,2017)]。受PseAAC成功处理蛋白质/肽序列的鼓舞,其思想和方法已扩展到处理DNA/RNA序列(陈等。, 2013,,2016年c;冯等。, 2017;林等。, 2014,2017年a,c(c);邱等。, 2014)通过PseKNC在计算基因组学中(Pse(磅/平方英寸)乌多K(K)-元组N个核苷酸C类合成)(陈等。2014年,2015). 根据一般PseAAC的概念(周,2011),任何蛋白质序列都可以作为PseAAC载体,由哪里T型是转置运算符,而整数是参数及其值以及组件将取决于如何从氨基酸序列中提取所需信息P(P),如下所述。 作为一种类型的一般心理交流(周,2011)GO(基因本体)已被广泛用于提高蛋白质亚细胞定位的预测质量(参见例如。万等。, 2013;吴等。, 2011;肖等。,2011年a,b条). 使用GO方法的优点是,映射到GO空间的蛋白质(而不是欧几里德空间或任何其他简单的几何空间)将更好地根据其亚细胞位置进行聚类,如(周和沈,2008). 关于使用GO方法预测蛋白质亚细胞定位的基本原理,以及对GO方法合理性的深入讨论,请参阅综合综述论文的第六节(周,2013).
然而,现有GO方法(参见示例。沈和周,2007;吴等。, 2011;肖等。,2011年a,b条)具有以下缺点。(i) 只有数字0和1(或其简单组合)用于合并GO信息,因此可能会遗漏一些重要信息。(ii)蛋白质载体的尺寸,即属于等式5,在之前的GO方法中非常高;例如1930年(周和蔡,2003),3043英寸(林等。, 2013)和9567英寸(周和沈,2006)从而可能导致高维灾难问题(王等。2008年).
这里,我们将介绍一种新的GO方法,通过该方法,我们可以通过筛选许多琐碎信息来掌握关键信息,从而显著降低PseAAC向量的维数等式5具体步骤如下。
第1步使用BLAST在Swiss-Prot数据库中搜索所有人类蛋白质,以查找与该蛋白质具有高同源性(即超过60%的配对序列一致性)的蛋白质P(P)属于公式4.由此获得的蛋白质被收集到一个子集中,,称为的同源集P(P)随后,检索中蛋白质的GO代码与P(P)每个GO代码都是一个包含7位数字的数字标签(参见示例。周和蔡,2003). 如果它根本没有GO代码,那么对中同源性第二高的蛋白质执行相同的操作; 如果它再次没有GO代码,那么对第三个最高同源的代码执行相同的操作;这样继续,直到获得一组go代码,如下所示哪里是k个-中蛋白质的第个GO代码根据上述顺序,首次发现了一组GO代码,以及是它具有的GO代码总数。假设我们从训练数据集中发现,具有与是牛顿(k),其中蛋白质的数量单位-第个子集是哪里是被调查的亚细胞位置总数(参见等式2或表1). 第2步。基于公式7,中的通用PseAAC矢量等式5其尺寸可以唯一定义为哪里N个(k个)是训练数据集中具有相同GO代码的人类蛋白质的总数运算符Max表示取不同值之间的最大值k个正是通过这种最大化操作来提取本研究中最重要的GO信息,并筛选出许多琐碎的GO代码来降低向量的维数。 在中列出补充材料S3PseAAC向量定义为等式8对于3106个序列不同的人类蛋白质补充材料S2分别是。如我们所见,当前PseAAC向量的维数已减少到14,比以前的方法小得多(周和蔡,2003;周和沈,2006;林等。, 2013).
2.3运算算法
五步规则中的第三步(周,2011)是关于运行预测的操作算法(或引擎)。在这里,我们采用了ML-GKR(多标签高斯核回归)分类器,如下所述。
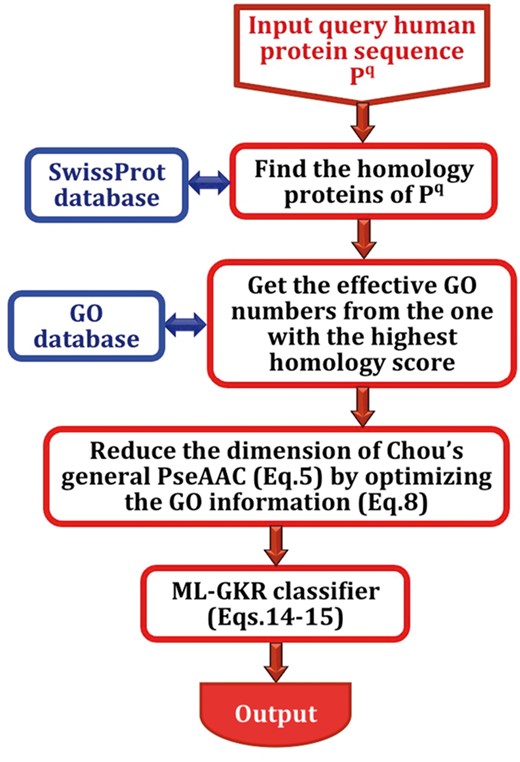
根据等式8或补充材料S3,的我-th人类蛋白在基准数据集中属于等式3可以表述为现在我们使用14-D矢量描述其在多标记系统中的亚细胞位置;即哪里同样,对于人类蛋白质的查询我们有其在多标记系统中的亚细胞位置标记应通过以下方式给出哪里这个在里面等式13由提供哪里N个(train)是用于训练模型的蛋白质的数量,是一个参数,其最佳值将在稍后确定,并且是平方欧氏距离(周和张,1995年)查询蛋白之间(等式12)和我-th蛋白(公式9)在基准数据集中; 即因此,位置标签向量属于等式13用于查询人类蛋白质定义明确,因此也可以明确预测其亚细胞位置。例如:如果而所有其他组件等式13等于 ,这意味着查询人类蛋白质位于第1、3和14亚细胞位置(参见。表1); 如果而所有其他人都是平等的 ,意味着查询蛋白仅位于第二亚细胞位置;等等。 通过上述程序开发的预测因子称为pLoc-mHum,其中“pLoc”代表“预测亚细胞定位”,“mHum”代表“多标签人类蛋白质”。如所示图1是说明pLoc-mHum如何工作的流程图。
3结果和讨论
如周的五步规则所述(周,2011)如何客观评估其预期准确性是开发新预测工具的重要步骤之一。为了解决这个问题,需要考虑两个问题。(i) 应该使用什么指标来定量反映预测者的质量?(ii)应采用什么测试方法来为指标打分?
3.1多标签系统的五个指标集
与用于测量单标签系统预测质量的度量不同,多标签系统的度量要复杂得多。为了让大多数实验科学家更直观、更容易理解,这里我们使用以下直观的周的五个指标(周,2013)最近被广泛用于研究各种多标签系统(参见例如。程等。2017a年,b条,c(c),d日,e(电子),(f),克;林等。, 2013;邱等。,2016年b;肖等。2017)以下为:哪里是查询蛋白质或测试蛋白质的总数,M(M)是被调查系统的不同标签总数(对于当前研究,是),是指操作其中集合以计算其元素数量的操作员,表示集合论中“联合”的符号,表示“十字路口”的符号,表示包含实验观察到的所有标签的子集k个-第个测试样品,表示包含为k个-第个样品,以及在等式17,带有上箭头的前四个指标被称为正度量,这意味着速率越大,预测质量就越好;带有向下箭头的第五个指标被称为负指标,意思正好相反。 发件人等式17我们可以看到以下内容:(i)由1定义的“瞄准”标准子方程用于检查正确预测标签与实际预测标签的比率或百分比;(ii)第二个子方程中定义的“覆盖率”用于检查相关系统中正确预测标签与实际标签的比率;(iii)3中的“准确性”第个子方程用于检查正确预测标签与总标签的平均比率,包括正确预测标签和错误预测标签以及预测中遗漏的真实标签;(iv)第四个子方程中的“绝对真”用于检查完全或完全正确的预测事件与总预测事件的比率;(v) 第五个子方程中的“绝对假”用于检查完全错误预测与总预测事件的比率。
3.2刀切试验
在统计预测中经常使用三种交叉验证方法。它们是:(i)独立数据集测试,(ii)二次抽样(或K折交叉验证)测试和(iii)折刀测试(周和张,1995年). 然而,在这三种测试中,折刀测试被认为是最不武断的测试,它总是能够为给定的基准数据集产生独特的结果,如(周,2011). 因此,折刀试验已得到广泛认可,并越来越多地被研究人员用于检查各种预测因子的质量(参见。Ali和Hayat,2015年;周和埃尔罗德,2003年;周和沈,2007b;埃斯马埃利等。, 2010;可汗等。, 2017;梅赫尔等。, 2017;穆罕默德等。, 2011;塔希尔和哈亚特,2016年;Zhou和Assa Munt,2001年). 因此,本研究中也使用了折刀试验。
3.3参数确定
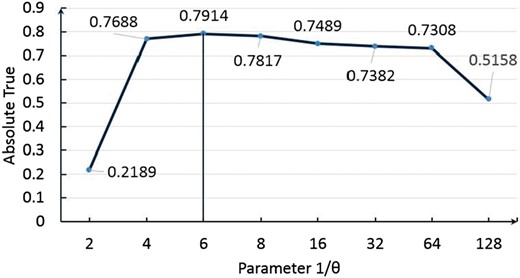
自等式15包含参数pLoc-mHum得到的预测结果将取决于参数的值。在本研究中通过最大化绝对真实率来确定(参见等式17)通过对基准数据集的jackknife验证。如所示图2,何时,绝对真实率达到最高分。该值将用于进一步研究。
图2。
一个图,显示了找到最优值的过程公式15中的值。更多解释请参见正文
3.4与最新预测值的比较
在中列出表2是当前pLoc-mHum预测值通过对基准数据集的折刀测试获得的速率(补充材料S1). 为了便于比较,该表中还列出了iLoc-Hum获得的相应结果(周等。, 2012),现有最强大的预测因子,用于识别具有单位点和多位点的人类蛋白质的亚细胞定位。
| 预测器. | 瞄准(b条. | 覆盖范围(b条. | 准确性(b条. | 绝对正确(b条. | 绝对错误(b条. |
|---|
| pLoc-mHum(位置-湿度)c(c) | 90.57% | 82.75% | 84.39% | 79.14% | 1.20% |
| iLoc-Hum公司d日 | 不适用 | 76.31% | 不适用 | 68.19% | 不适用 |
| 预测器. | 瞄准(b条. | 覆盖范围(b条. | 准确性(b条. | 绝对正确(b条. | 绝对错误(b条. |
|---|
| pLoc-mHum(位置-湿度)c(c) | 90.57% | 82.75% | 84.39% | 79.14% | 1.20% |
| iLoc-Hum公司d日 | 不适用 | 76.31% | 不适用 | 68.19% | 不适用 |
| 预测器. | 瞄准(b条. | 覆盖范围(b条. | 准确性(b条. | 绝对正确(b条. | 绝对错误(b条. |
|---|
| pLoc mHum基因c(c) | 90.57% | 82.75% | 84.39% | 79.14% | 1.20% |
| iLoc-Hum公司d日 | 不适用 | 76.31% | 不适用 | 68.19% | 不适用 |
| 预测器. | 瞄准(b条. | 覆盖范围(b条. | 准确性(b条. | 绝对正确(b条. | 绝对错误(b条. |
|---|
| pLoc-mHum(位置-湿度)c(c) | 90.57% | 82.75% | 84.39% | 79.14% | 1.20% |
| iLoc-Hum公司d日 | 不适用 | 76.31% | 不适用 | 68.19% | 不适用 |
如所示表2,在等式17用于定量测量多标签预测器的质量(周,2013)iLoc-Hum的“瞄准”、“准确度”和“绝对错误”率(周等。, 2012)缺少,表明在检查预测质量方面缺乏严谨性。换句话说,iLoc-Hum的作者只报告了“覆盖率”和“绝对真实”的比率。但这两种方法都显著低于本文提出的电流预测器pLoc-mHum所达到的相应速率。正如全面审查中指出的那样(周,2013),在中列出的上述五个指标中表2,最重要的是“绝对正确”和“绝对错误”。多标签预测器很难提高其绝对正确率,降低其绝对错误率。因此,在开发预测单位点和多位点蛋白质亚细胞定位的方法时,许多研究人员甚至没有提到“绝对正确”和“绝对错误”率。与此相反,可以从表2当前pLoc-mHum预测值的绝对真实率比iLoc-Hum预测值高出10%以上,明显表明其优越性。
在中列出表3是pLoc-Hum针对14个亚细胞位置中的每个位置获得的相应结果。从表中可以看出,14个亚细胞位置中的每个位置的得分也很高,这与报告中的整体表现完全一致表2.
| 我. | 位置一. | 锡(i)b条. | Sp(i)b条. | 账户(i)b条. | MCC(i)b条. |
|---|
| 1 | 中心体 | 0.7143 | 0.9937 | 0.9868 | 0.7219 |
| 2 | 细胞质 | 0.7760 | 0.9415 | 0.8979 | 0.7322 |
| 三 | 细胞骨架 | 0.7722 | 0.9970 | 0.9913 | 0.8159 |
| 4 | 内质网 | 0.7817 | 0.9864 | 0.9713 | 0.7857 |
| 5 | 内吞体 | 0.4583 | 0.9984 | 0.9942 | 0.5586 |
| 6 | 细胞外 | 0.8390 | 0.9838 | 0.9659 | 0.8400 |
| 7 | 高尔基体 | 0.7453 | 0.9871 | 0.9746 | 0.7390 |
| 8 | 溶酶体 | 0.8831 | 0.9954 | 0.9926 | 0.8520 |
| 9 | 微粒体 | 0.7083 | 0.9971 | 0.9948 | 0.6780 |
| 10 | 线粒体 | 0.9258 | 0.9865 | 0.9794 | 0.9017 |
| 11 | 核心 | 0.8580 | 0.9242 | 0.9024 | 0.7797 |
| 12 | 过氧化物酶体 | 0.8511 | 0.9974 | 0.9952 | 0.8397 |
| 13 | 质膜 | 0.8249 | 0.9815 | 0.9636 | 0.8175 |
| 14 | 突触 | 0.7727 | 0.9994 | 0.9977 | 0.8304 |
| 我. | 位置一. | 锡(i)b条. | Sp(i)b条. | 账户(i)b条. | MCC(i)b条. |
|---|
| 1 | 中心体 | 0.7143 | 0.9937 | 0.9868 | 0.7219 |
| 2 | 细胞质 | 0.7760 | 0.9415 | 0.8979 | 0.7322 |
| 三 | 细胞骨架 | 0.7722 | 0.9970 | 0.9913 | 0.8159 |
| 4 | 内质网 | 0.7817 | 0.9864 | 0.9713 | 0.7857 |
| 5 | 内吞体 | 0.4583 | 0.9984 | 0.9942 | 0.5586 |
| 6 | 细胞外 | 0.8390 | 0.9838 | 0.9659 | 0.8400 |
| 7 | 高尔基体 | 0.7453 | 0.9871 | 0.9746 | 0.7390 |
| 8 | 溶酶体 | 0.8831 | 0.9954 | 0.9926 | 0.8520 |
| 9 | 微粒体 | 0.7083 | 0.9971 | 0.9948 | 0.6780 |
| 10 | 线粒体 | 0.9258 | 0.9865 | 0.9794 | 0.9017 |
| 11 | 核心 | 0.8580 | 0.9242 | 0.9024 | 0.7797 |
| 12 | 过氧化物酶体 | 0.8511 | 0.9974 | 0.9952 | 0.8397 |
| 13 | 质膜 | 0.8249 | 0.9815 | 0.9636 | 0.8175 |
| 14 | 突触 | 0.7727 | 0.9994 | 0.9977 | 0.8304 |
| 我. | 位置一. | 锡(i)b条. | Sp(i)b条. | 账户(i)b条. | MCC(i)b条. |
|---|
| 1 | 中心体 | 0.7143 | 0.9937 | 0.9868 | 0.7219 |
| 2 | 细胞质 | 0.7760 | 0.9415 | 0.8979 | 0.7322 |
| 三 | 细胞骨架 | 0.7722 | 0.9970 | 0.9913 | 0.8159 |
| 4 | 内质网 | 0.7817 | 0.9864 | 0.9713 | 0.7857 |
| 5 | 内吞体 | 0.4583 | 0.9984 | 0.9942 | 0.5586 |
| 6 | 细胞外 | 0.8390 | 0.9838 | 0.9659 | 0.8400 |
| 7 | 高尔基体 | 0.7453 | 0.9871 | 0.9746 | 0.7390 |
| 8 | 溶酶体 | 0.8831 | 0.9954 | 0.9926 | 0.8520 |
| 9 | 微粒体 | 0.7083 | 0.9971 | 0.9948 | 0.6780 |
| 10 | 线粒体 | 0.9258 | 0.9865 | 0.9794 | 0.9017 |
| 11 | 核心 | 0.8580 | 0.9242 | 0.9024 | 0.7797 |
| 12 | 过氧化物酶体 | 0.8511 | 0.9974 | 0.9952 | 0.8397 |
| 13 | 质膜 | 0.8249 | 0.9815 | 0.9636 | 0.8175 |
| 14 | 突触 | 0.7727 | 0.9994 | 0.9977 | 0.8304 |
| 我. | 位置一. | 锡(i)b条. | Sp(i)b条. | 会计科目(i)b条. | MCC(i)b条. |
|---|
| 1 | 中心体 | 0.7143 | 0.9937 | 0.9868 | 0.7219 |
| 2 | 细胞质 | 0.7760 | 0.9415 | 0.8979 | 0.7322 |
| 三 | 细胞骨架 | 0.7722 | 0.9970 | 0.9913 | 0.8159 |
| 4 | 内质网 | 0.7817 | 0.9864 | 0.9713 | 0.7857 |
| 5 | 内吞体 | 0.4583 | 0.9984 | 0.9942 | 0.5586 |
| 6 | 细胞外 | 0.8390 | 0.9838 | 0.9659 | 0.8400 |
| 7 | 高尔基体 | 0.7453 | 0.9871 | 0.9746 | 0.7390 |
| 8 | 溶酶体 | 0.8831 | 0.9954 | 0.9926 | 0.8520 |
| 9 | 微粒体 | 0.7083 | 0.9971 | 0.9948 | 0.6780 |
| 10 | 线粒体 | 0.9258 | 0.9865 | 0.9794 | 0.9017 |
| 11 | 核心 | 0.8580 | 0.9242 | 0.9024 | 0.7797 |
| 12 | 过氧化物酶体 | 0.8511 | 0.9974 | 0.9952 | 0.8397 |
| 13 | 质膜 | 0.8249 | 0.9815 | 0.9636 | 0.8175 |
| 14 | 突触 | 0.7727 | 0.9994 | 0.9977 | 0.8304 |
3.5 Web服务器和用户指南
如中所述(周和沈,2009)用户友好且可公开访问的网络服务器代表了开发更实用的预测器或任何计算工具的未来方向。实际上,用户友好的网络服务器,如最近一系列出版物所示(陈等。,2016年b,2017;程等。,2017年a;贾等。,2016年a,c(c),d日;线路接口单元等。2017b年,d日;邱等。2017a年,b条;邱等。,2016年a,c(c);肖等。, 2016;徐等。, 2013,2014,2017;张等。, 2016)将显著提高理论工作的影响,因为它们可以吸引广泛的实验科学家(周,2015). 因此,建立了pLoc-mHum预测器的网络服务器。此外,为了最大限度地提高用户的便利性,在补充材料S4.
4结论
人类蛋白质亚细胞位置预测是一个具有挑战性的问题,特别是当查询的人类蛋白质具有多标签特征时,这意味着它们可能发生在两个或多个不同的位置。在这里,我们通过将最优GO信息合并到Chou的一般PseAAC中,开发了一种新的预测因子pLoc-mHum(周,2011). 与iLoc-Hum相比(周等。, 2011)作为现有最强大的预测因子,它还具有处理人类蛋白质多位置的能力,根据广泛用于衡量多标签预测因子质量的指标,新预测因子获得的分数明显优于iLoc-Hum。
为什么新的预测工具如此强大?其本质是,在新方法中有两个重要的方程;即等式8和15通过前者,通过剔除关键特征,可以大幅降低一般PseAAC向量的维数,从而显著降低大量噪声。通过后者,可以以逻辑和一致的方式自然地导出多个位置,而不是像大多数现有方法那样通过人工插入阈值或截止值。
由于可公开访问的web服务器代表了开发更实用的预测方法的未来方向(周和沈,2009),pLoc-mHum的网络服务器已经建立,其用户指南见补充材料S4预计pLoc-mHum将成为一种非常有用的高通量工具,用于注释人类蛋白质的亚细胞位置。
致谢
作者希望感谢三位匿名审稿人,他们的建设性意见对加强本文的介绍非常有帮助。
基金
本项工作得到了国家自然科学基金(No.31560316,61261027,61262038,61202313,31260273)、江西省国家自然科学项目(No.20132BAB201053)和江西省对外科技合作项目(No.20120BDH80023)的资助,江西省教育厅(GJJ160866)。本论文得到了国家自然科学基金(61271114号和61203325号)和上海市教育委员会创新计划(14ZZ068号)的部分资助。
利益冲突:未声明。
参考文献
艾哈迈德
美国。
等(
2015
)通过将二肽组分纳入Chou的一般PseAAC中来鉴定热休克蛋白家族和J蛋白类型
.计算。生物识别方法程序
.,122
,165
–174
.
阿里
F、。
,海亚特
M。
(
2015
)基于投票特征区间和周伪氨基酸组成的膜蛋白类型分类
.J.西奥。生物
.,384
,78
–83
.
蔡
年月日。
,周
K.C.公司。
(
2000
)用神经网络预测原核和真核蛋白质的亚细胞位置
.分子细胞生物学。Res.Commun公司
.,4
,172
–173
.
曹
国防部。
等(
2013
)propy:生成Chou的PseAAC的各种模式的工具
.生物信息学
,29
,960
–962
.
塞达诺
J。
等(
1997
)蛋白质氨基酸组成与细胞定位的关系
.分子生物学杂志
.,266
,594
–600
.
陈
J。
等(
2007
)用氨基酸对抗原性量表预测线性B细胞表位
.氨基酸
,33
,423
–428
.
陈
J。
等(
2016年a
)dRHP-PseRA:使用基于轮廓的伪蛋白序列和秩聚集检测远程同源蛋白
.科学。代表
.,6
,32333
.
陈
西。
等(
2016年b
)iACP:一种基于序列的抗癌肽鉴定工具
.肿瘤靶点
,7
,16895
–16909
.
陈
西。
等(
2016年c
)iRNA-PseU:鉴定RNA假尿苷位点
.分子治疗核酸
,5
,e332(电子332)
.
陈
西。
等(
2017
)iRNA-AI:识别RNA序列中腺苷到肌苷的编辑位点
.肿瘤靶点
,8
,4208
–4217
.
陈
西。
等(
2013
)iRSpot-PseDNC:识别假二核苷酸组成的重组点
.核酸研究
.,41
,e68页。
陈
西。
等(
2014
)PseKNC:生成伪K元组核苷酸组成的灵活网络服务器
.分析。生物化学
.,456
,53
–60
.
陈
西。
等(
2015
)伪核苷酸组成或PseKNC:分析基因组序列的有效公式
.分子生物系统
.,11
,2620
–2634
.
程
十、。
等(
2017年b
)iATC-mISF:一种预测解剖治疗化学物质类别的多标签分类器
.生物信息学
,33
,341
–346
.
程
十、。
等(
2017年c
)pLoc-mPlant:通过将最佳GO信息合并到通用PseAAC中来预测多位置植物蛋白的亚细胞定位
.分子生物系统
.,13
,1722
–1727
.
程
十、。
等(
2017天
)pLoc-mVirus:通过将最佳GO信息合并到通用PseAAC中来预测多位置病毒蛋白的亚细胞定位
.基因
,628
,315
–321
.
周
K.C.公司。
(
2000
)结合准序列效应预测蛋白质亚细胞位置
.生物化学。生物物理学。Res.Comm.(英国广播公司)
,278
,477
–483
.
周
K.C.公司。
(
2001
)利用伪氨基酸组成预测蛋白质细胞属性
.蛋白质结构。功能。遗传学。(勘误表:同上,2001年,第44卷,第60页)
,43
,246
–255
.
周
K.C.公司。
(
2005
)利用两亲性伪氨基酸组成预测酶亚家族类别
.生物信息学
,21
,10
–19
.
周
K.C.公司。
(
2009
)伪氨基酸组成及其在生物信息学、蛋白质组学和系统生物学中的应用
.货币。蛋白质组学
,6
,262
–274
.
周
K.C.公司。
(
2011
)蛋白质属性预测和伪氨基酸组成的一些评论(50周年回顾)
.J.西奥。生物
.,273
,236
–247
.
周
K.C.公司。
(
2013
)分子生物系统中多标签属性预测的几点注记
.分子生物晶体
.,9
,1092
–1100
.
周
K.C.公司。
(
2015
)生物信息学对药物化学的影响
.药物化学
.,11
,218
–234
.
周
K.C.公司。
(
2017
)生物科学进步推动药物化学的空前革命
.货币。顶部。药物化学
.,17
,2358
周
K.C.公司。
,蔡
Y.D.年。
(
2002
)利用功能域组成和支持向量机预测蛋白质亚细胞位置
.生物学杂志。化学
.,277
,45765
–45769
.
周
K.C.公司。
,蔡
Y.D.年。
(
2003
)结合基因本体预测蛋白质亚细胞定位的新混合方法
.生物化学。生物物理学。Res.Commun公司。(英国广播公司)
,311
,743
–747
.
周
K.C.公司。
,埃尔罗德
D.W.公司。
(
1998
)用判别函数预测原核蛋白的亚细胞定位
.生物化学。生物物理学。Res.Commun公司。(英国广播公司)
,252
,63
–68
.
周
K.C.公司。
,埃尔罗德
D.W.公司。
(
1999年a
)膜蛋白类型和亚细胞位置的预测
.蛋白质结构。功能。基因
.,34
,137
–153
.
周
K.C.公司。
,埃尔罗德
D.W.公司。
(
1999年b
)蛋白质亚细胞定位预测
.蛋白质工程
.,12
,107
–118
.
周
K.C.公司。
,埃尔罗德
D.W.公司。
(
2003
)酶家族分类的预测
.蛋白质组研究杂志
.,2
,183
–190
.
周
K.C.公司。
,沈
H.B.公司。
(
2006
)融合优化的证据理论K-最近邻分类器预测真核蛋白亚细胞定位
.蛋白质组研究杂志
.,5
,1888
–1897
.
周
K.C.公司。
,沈
H.B.公司。
(
2007年a
)Euk-mPLoc:一种融合多位点的大规模真核蛋白亚细胞定位预测融合分类器
.蛋白质组研究杂志
.,6
,1728
–1734
.
周
K.C.公司。
,沈
H.B.公司。
(
2007年b
)MemType-2L:一个通过Pse-PSSM整合进化信息来预测膜蛋白及其类型的网络服务器
.生物化学。生物物理学。Res.Commun公司。(英国广播公司)
,360
,339
–345
.
周
K.C.公司。
,沈
H.B.公司。
(
2007年c
)蛋白质亚细胞定位预测研究进展
.分析。生物化学
.,370
,1
–16
.
周
K.C.公司。
,沈
H.B.公司。
(
2008
)Cell-PLoc:一个用于预测各种生物体中蛋白质亚细胞定位的Web服务器包(更新版本:Cell-PLoc 2.0:一个改进的用于预测各种有机体中蛋白质亚胞定位的网络服务器包,自然科学,2010,21090–1103)
.自然协议
.,三
,153
–162
.
周
K.C.公司。
,沈
H.B.公司。
(
2009
)开发用于预测蛋白质属性的网络服务器的最新进展
.自然科学
.,01
,63
–92
.
周
K.C.公司。
,沈
H.B.公司。
(
2010年a
)预测单位点和多位点真核蛋白亚细胞定位的新方法:Euk-mPLoc 2.0
.公共科学图书馆
,5
,电子9931
.
周
K.C.公司。
,沈
H.B.公司。
(
2010年b
)Plant-mPLoc:一种自顶向下的策略,用于增强预测植物蛋白质亚细胞定位的能力
.公共科学图书馆
,5
,电子11335
.
周
K.C.公司。
等(
2011
)iLoc-Euk:一种用于预测单重和多重真核蛋白亚细胞定位的多标记分类器
.公共科学图书馆一号
,6
,e18258(电子18258)
.
周
K.C.公司。
等(
2012
)iLoc-Hum:使用累加-标签尺度预测人类蛋白质的亚细胞位置,包括单位点和多位点
.分子生物晶体
.,8
,629
–641
.
周
K.C.公司。
,张
C.T.公司。
(
1995
)综述:蛋白质结构类的预测
.批评。生物化学版。分子生物学
.,30
,275
–349
.
丁
Y.S.公司。
,张
T.L.公司。
(
2008
)利用Chou的伪氨基酸组成预测凋亡蛋白的亚细胞定位:基于免疫遗传算法的集成分类器方法
.模式识别。莱特
.,29
,1887
–1892
.
杜
第页。
等(
2012
)PseAAC-Builder:一个跨平台独立程序,用于生成各种特殊的Chou伪氨基酸成分
.分析。生物化学
.,425
,117
–119
.
杜
Q.S.(质量标准)。
等(
2017
)2L-PCA:一种用于药物定量设计的两级主成分分析仪及其应用
.肿瘤靶点
,8
,70564
–70578
.
埃马努埃尔松
O。
等(
2000
)基于N端氨基酸序列预测蛋白质的亚细胞定位
.分子生物学杂志
.,300
,1005
–1016
.
埃斯马埃利
M。
等(
2010
)利用周伪氨基酸组成的概念预测人乳头瘤病毒的风险类型
.J.西奥。生物
.,263
,203
–209
.
冯
第页。
等(
2017
)iRNA-PseColl:通过将核苷酸的集体效应纳入PseKNC来识别不同RNA修饰的发生位置
.分子治疗核酸
,7
,155
–163
.
加迪
法学博士。
(
2003
)PSORT-B:改进革兰氏阴性菌蛋白质亚细胞定位预测
.核酸研究
.,31
,3613
–3617
.
荣耀
E.公司。
,墨菲
射频。
(
2007
)自动亚细胞定位和高通量显微镜
.开发单元
.,12
,7
–16
.
黄
C。
,元
J。
(
2013
)利用Chou伪氨基酸组成的一般形式的径向基函数和PSSM预测单位点和多位点蛋白质的亚细胞位置
.生物系统
,113
,50
–57
.
贾
J。
等(
2016年a
)iCar-PseCp:通过蒙特卡罗采样确定蛋白质中的羰基化位点,并将序列耦合效应纳入一般PseAAC
.肿瘤靶点
,7
,34558
–34570
.
贾
J。
等(
2016年b
)通过将物理化学性质和平稳小波变换结合到伪氨基酸组成中来识别蛋白质结合位点(iPPBS-PseAAC)
.《生物分子杂志》。结构。动态。(JBSD)
,34
,1946
–1961
.
贾
J。
等人(
2016年c
)pSuc-Lys:使用PseAAC和集成随机森林方法预测蛋白质中的赖氨酸琥珀酰化位点
.J.西奥。生物
.,394
,223
–230
.
贾
J。
等(
2016年d
)pSumo-CD:通过将序列耦合效应纳入一般PseAAC,使用协方差判别算法预测蛋白质中的sumoylation位点
.生物信息学
,32
,3133
–3141
.
可汗
M。
等(
2017
)Unb-DPC:通过将非偏倚二肽成分纳入Chou的一般PseAAC中来识别分枝杆菌膜蛋白类型
.J.西奥。生物
.,415
,13
–19
.
林
H。
等(
2014
)iPro54-PseKNC:一种基于序列的预测因子,用于识别具有伪k元组核苷酸组成的原核生物sigma-54启动子
.核酸研究
.,42
,12961
–12972
.
林
S.X.公司。
,拉波因特
J。
(
2013
)理论生物学和实验生物学合二为一——纪念郭振洲教授50周年和Richard Giegé教授40周年的研讨会
.J.生物识别。科学。工程(JBiSE)
,06
,435
–442
.
林
W.Z.公司。
等(
2013
)iLoc-Animal:预测动物蛋白质亚细胞定位的多标记学习分类器
.分子生物系统
.,9
,634
–644
.
线路接口单元
B。
等(
2017年a
)iRSpot-EL:用集成学习方法识别重组点
.生物信息学
,33
,35
–41
.
线路接口单元
B。
等(
2017年b
)Pse-Analysis:基于伪成分和核方法的DNA/RNA和蛋白质/肽序列分析的python包
.肿瘤靶点
,8
,13338
–13343
.
线路接口单元
B。
等(
2017年c
)2L-piRNA:一种用于识别piwi相互作用RNA及其功能的双层集成分类器
.分子治疗核酸
,7
,267
–277
.
线路接口单元
L.M.有限公司。
等(
2017天
)iPGK-PseAAC:通过将四个不同层次的氨基酸成对偶联信息合并到一般PseAAA中来识别蛋白质中的赖氨酸磷酸甘油化位点
.药物化学
.,13
,552
–559
.
梅赫尔
P.K.公司。
等(
2017
)通过将组成、物理化学和结构特征纳入Chou的一般PseAAC,预测抗菌肽的准确性得到提高
.科学。代表
.,7
,42362
.
梅
美国。
(
2012
)基于多标记同源知识转移学习的Chou的PseAAC公式预测植物蛋白质亚细胞多定位
.J.西奥。生物
.,310
,80
–87
.
穆罕默德
H。
等(
2011
)基于Chou伪氨基酸组成和支持向量机的GABA(A)受体蛋白预测
.J.西奥。生物
.,281
,18
–23
.
穆罕默德·贝吉
M。
等(
2011
)基于周伪氨基酸组成概念的金属蛋白酶家族机器学习预测
.J.结构。功能。基因组学
,12
,191
–197
.
蒙道
美国。
,派
P.P.公司。
(
2014
)周的伪氨基酸成分改进了基于序列的抗冻蛋白预测
.J.西奥。生物
.,356
,30
–35
.
Nakai公司
英国。
(
2000
)蛋白质分选信号与亚细胞定位预测
.高级蛋白质化学
.,54
,277
–344
.
南尼
L。
,鲁米尼
答:。
(
2008
)遗传编程创建周氏伪氨基酸特征用于亚线粒体定位
.氨基酸
,34
,653
–660
.
纳尼
L。
等(
2012
)通过融合一组基于周氏伪氨基酸组成变体和进化信息的分类器来识别细菌毒性蛋白
.IEEE-ACM传输。计算。生物信息素
.,9
,467
–475
.
Pacharawongsakda公司
E.公司。
,Theeramunkong公司
T。
(
2013
)通过半监督学习和Chou’s PseAAC降维通用模型预测单复合体和复合蛋白的亚细胞位置
.IEEE传输。纳米生物学
.,12
,311
–320
.
邱
W.R.公司。
等(
2017年b
)iRNAm5C-PseDNC:通过将物理化学性质纳入伪二核苷酸组成来识别RNA 5-甲基胞嘧啶位点
.肿瘤靶点
,8
,41178
–41188
.
邱
W.R.公司。
等(
2016年a
)iHyd-PseCp:通过将序列偶联效应纳入一般PseAAC来识别蛋白质中的羟脯氨酸和羟赖氨酸
.肿瘤靶点
,7
,44310
–44321
.
邱
W.R.公司。
等(
2016年b
)iPTM-mLys:鉴定多个赖氨酸PTM位点及其不同类型
.生物信息学
,32
,3116
–3123
.
邱
W.R.公司。
等(
2016年c
)iPhos-PseEn:通过将不同的伪成分融合到集成分类器中来识别蛋白质中的磷酸化位点
.肿瘤靶点
,7
,51270
–51283
.
邱
W.R.公司。
等(
2014
)iRSpot TNCPseAAC:识别具有三核苷酸组成和伪氨基酸成分的重组点
.国际分子科学杂志。(IJMS)
,15
,1746
–1766
.
拉希米
M。
等(
2017
)OOgenesis_Pred:一种基于序列的方法,通过六种不同模式的Chou伪氨基酸组成预测卵子发生蛋白
.J.西奥。生物
.,414
,128
–136
.
莱因哈特
答:。
,哈伯德
T。
(
1998
)利用神经网络预测蛋白质的亚细胞位置
.核酸研究
.,26
,2230
–2236
.
萨胡
S.S.公司。
,熊猫
G.公司。
(
2010
)基于Chou伪氨基酸组成的蛋白质结构类预测特征表示新方法
.计算。生物化学
.,34
,320
–327
.
沈
H.B.公司。
,周
K.C.公司。
(
2007
)Hum-mPLoc:一种集成分类器,通过合并多个位点的样本进行大规模人类蛋白质亚细胞位置预测
.生物化学。生物物理学。Res.Commun公司。(英国广播公司)
,355
,1006
–1011
.
沈
H.B.公司。
,周
K.C.公司。
(
2009
)一种自顶向下的方法来增强预测人类蛋白质亚细胞定位的能力:Hum-mPLoc 2.0
.分析。生物化学
.,394
,269
–274
.
沈
H.B.公司。
,周
K.C.公司。
(
2010年a
)Gneg-mPLoc:一种自顶向下的策略,用于提高预测革兰氏阴性细菌蛋白质亚细胞定位的质量
.J.西奥。生物
.,264
,326
–333
.
沈
H.B.公司。
,周
K.C.公司。
(
2010年b
)Virus-mPLoc:一种融合多位点的病毒蛋白亚细胞定位融合分类器
.《生物分子杂志》。结构。动态。(JBSD)
,28
,175
–186
.
塔希尔
M。
,海亚特
M。
(
2016
)iNuc-STNC:通过扩展SAAC和Chou的PseAAC概念识别基因组中核小体定位的基于序列的预测因子
.分子生物晶体
.,12
,2587
–2593
.
特里帕西
第页。
,潘迪
邮政编码:。
(
2017
)结合谱图聚类和Chou伪氨基酸组成的无比对蛋白质折叠类型分类新方法
.J.西奥。生物
.,424
,49
–54
.
万
美国。
等人(
2013
)GOASVM:通过将术语频率基因本体纳入周氏伪氨基酸组成的一般形式,实现亚细胞位置预测
.J.西奥。生物
.,323
,40
–48
.
王
T。
等(
2008
)用LLDA算法预测膜蛋白类型
.蛋白质肽Lett
.,15
,915
–921
.
王
十、。
等(
2013
)病毒-ECC-mPLoc:基于Chou伪氨基酸组成的一般形式预测单位点和多位点病毒蛋白亚细胞定位的多标记预测因子
.蛋白质肽Lett
.,20
,309
–317
.
吴
Z.C.公司。
等(
2011
)iLoc-Plant:一种预测植物单位点和多位点蛋白质亚细胞定位的多标记分类器
.分子生物学系统
.,7
,3287
–3297
.
吴
Z.C.公司。
等(
2012
)iLoc-Gpos:预测单复合体和多重革兰氏阳性细菌蛋白亚细胞定位的多层分类器
.蛋白质肽Lett
.,19
,4
–14
.
肖
十、。
等(
2013
)iAMP-2L:一种用于鉴定抗菌肽及其功能类型的两级多标签分类器
.分析。生物化学
.,436
,168
–177
.
肖
十、。
等(
2011年a
)iLoc-Virus:一种用于识别单位点和多位点病毒蛋白亚细胞定位的多标记学习分类器
.J.西奥。生物
.,284
,42
–51
.
肖
十、。
等(
2011年b
)一种用于预测单位点和多位点革兰氏阴性细菌蛋白亚细胞定位的多标记分类器
.公共科学图书馆
,6
,e20592号
.
肖
十、。
等(
2016
)iROS-gPseKNC:通过将二核苷酸位置特异性倾向纳入一般伪核苷酸组成来预测DNA中的复制起始位点
.肿瘤靶点
,7
,34180
–34189
.
肖
十、。
等(
2017
)pLoc-mGpos:将关键基因本体信息合并到通用PseAAC中,用于预测革兰氏阳性细菌蛋白质的亚细胞定位
.自然科学
.,9
,331
–349
.
徐
年。
等(
2017
)iPreny-PseAAC:通过将两层序列偶联到PseAAC中来识别蛋白质中的C端半胱氨酸丙烯化位点
.药物化学
.,13
,544
–551
.
徐
年。
等(
2013
)iSNO-AAPair:将氨基酸成对偶联到PseAAC中预测蛋白质中半胱氨酸S-亚硝基化位点
.同行J
,1
,e171。
徐
年。
等(
2014
)iHyd-PseAAC:通过将二肽位置特异性倾向纳入伪氨基酸组成来预测蛋白质中的羟脯氨酸和羟赖氨酸
.国际分子科学杂志
.,15
,7594
–7610
.
张
C.J.公司。
等(
2016
)iOri-Human:通过将二核苷酸的物理化学性质纳入伪核苷酸组成来确定人类复制起源
.肿瘤靶点
,7
,69783
–69793
.
钟
W.Z.公司。
,周
S.F.公司。
(
2014
)药物开发和生物医学的分子科学
.国际分子科学杂志
.,15
,20072
–20078
.
周
G.P.公司。
,阿萨姆
N。
(
2001
)蛋白质结构类预测的一些见解
.蛋白质结构。功能。基因
.,44
,57
–59
.
周
G.P.公司。
,钟
W.Z.公司。
(
2016
)药物化学展望
.货币。顶部。药物化学
.,16
,381
–382
.
周
X.B.公司。
等(
2007
)利用Chou的两亲性伪氨基酸组成和支持向量机预测酶亚科类别
.J.西奥。生物
.,248
,546
–551
.
©作者2017。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oup.com








{kind=link}
{kind=link}