摘要
动机:DNA序列数据的快速增长产生了一种需要,即需要找到图形工具来以可感知的形式研究DNA序列。然而,很难在一个图中处理以下问题:(i)退化,(ii)信息丢失,(iii)在多维图中观察的困难,(iv)表示长DNA序列时的可视化困难,以及(v)需要反映有用的信息。
结果:双矢量曲线(DV-Curve,Dual Vector Curve)用两个矢量表示一个DNA序列字母表,不仅避免了信息的退化和丢失,而且无论序列是否长,都具有良好的可视化效果,能够反映DNA序列的长度。详细介绍了DV-Curve在突变分析和两种相似性分析中的应用。DV曲线是一种有意义的工具,生物学家可以通过它找到有用的生物学知识。
可利用性:有关DV-Curve的相应软件,请访问http://bmchust.3322.org/Data/Soft/332-DV-Curve2.0.zip
联系人: zhangzhujin@gmail.com
1简介
DNA数据库中DNA序列数据的快速增长使得DNA的表示对于有效地整理、组织、识别、检索和搜索序列数据至关重要。一些调查人员,例如Hamori(1989),杰弗里(1990)和南迪(1994),考虑DNA序列的图形表示。这种表示法的优点是可以对数据进行视觉检查,有助于识别、搜索、组织和分析DNA序列。南迪(1994)通过将A(腺嘌呤)、G(鸟嘌呤x个), (+x个), (−年)和(+年)分别沿着笛卡尔坐标轴的正轴和负轴,然后是构成所考虑序列的基,我们将其与坐标系的遍历积分点相关联。
但这种DNA的表示伴随着(i)与结果曲线本身的交叉和重叠相关的信息丢失;以及(ii)电路产生的简并。因此,一些学者对Nandy的二维图形模型进行了改进。例如,郭等。(2001)创建低简并度模型;和Wu等。(2003)设计了一种非简并表示。然而,两者都不能同时处理这两个问题。
为了彻底解决这两个问题,引入了多维图形表示,例如3D(Hamori和Ruskin,1983; 齐和范,2007; 气等。,2007; 张和张,1994; 张等。,2003),4D(池和丁,2005)和6D(廖和王,2004)在以后的研究中。然而,所有这些多维方法都很难可视化,因为很难知道多维图形中一个点的每个轴的确切值,并且您不知道在您可以看到的点后面是否存在另一个点。
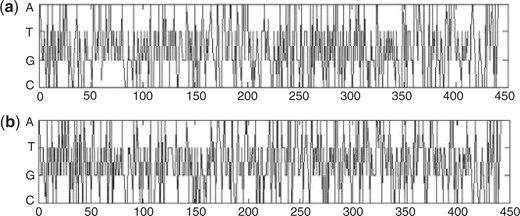
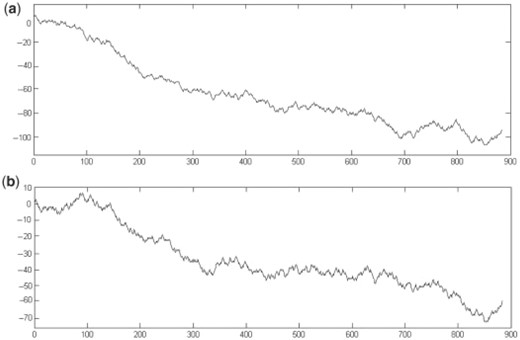
因此,一些精英回归2D图形表示。兰迪奇等。(2003年a)、姚明和王(2004)还有气和气(2007)创建了没有退化和没有信息丢失的2D图形表示。然而,从图1,很难确定图1a和b是不同的。因为当DNA序列大于300 bp时,这些表征的可视化也变得困难。
图1。
人类β-珠蛋白基因完整编码序列的二维图形表示(一)和负鼠(b条)(Randić)的等。,2003年a). 很难确定(a)和(b)中的序列不同。因为当DNA序列大于300 bp时,这种方法的可视化变得困难。
这里,没有退化和信息丢失的DV-Curve(双矢量曲线)具有良好的可视性来表示长序列。它也很简单,可以反映DNA序列的长度。详细介绍了DV-Curve的三个应用。
2 DV-曲线
在本节中,我们用生动的图形描述DV-Curve的构造。读者可以很容易地理解DV-Curve是如何由图2根据DV曲线的构造,提出了两个数学模型。第一个是“从DNA序列到DV曲线”,第二个是“由DV曲线到DNA序列”。最后,利用数学方法和实验结果,我们可以证明DV-Curve的几个高级特性。
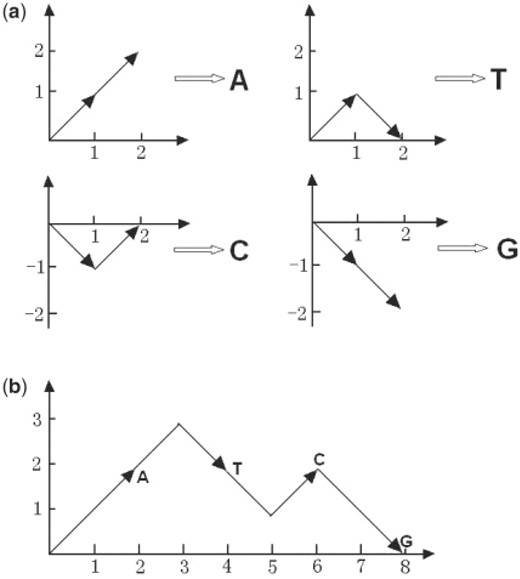
图2。
(一)DV-Curve的四个核苷酸A、T、C和G的表示。(b条)序列“ATCG”的DV-Curve。
2.1 DV-曲线的构造
在本小节中,给出了DV-Curve的构造。如所示图2a、 a、T、C和G的每个字母表由两个向量表示,如下所示: 可以获得DV-Curve(例如,图2b) 通过逐个连接所有矢量。
因此,我们可以从构造中了解DV-Curve的一些特性:(i)DV-Curve's very simple and need to set parameters。(ii)DV-曲线沿延伸2 UX(X)代表每个核苷酸的轴,无论A、T、C或G。(iii)DV-曲线不需要沿添加任何内容Y(Y)轴代表T和C,而需要添加2 U代表A,添加-2 U代表G。(iv)DV-曲线具有很好的对称性。A和G是对称的;T和C也是对称的。(v) 如果端点的值沿Y(Y)axis大于零,这意味着DNA序列中的A多于G。如果端点的值沿Y(Y)轴小于零,这意味着DNA序列中G多于A。
2.2 DV曲线的数学模型
根据DV曲线的构造,我们在本小节中提出了两个DV曲线数学模型。首先,我们需要定义一些在整篇文章中有效的常见描述和变量:模型1:给定DNA序列,绘制DV-Curve 我们将DNA序列描述为序列=S公司1S公司2…S公司我…S公司n个哪里S公司我∈ {A类,T型,C类,G公司},n个是这个DNA序列的长度序列是整个DNA序列。这意味着DNA序列序列由许多字母连接。
(X(X)j个,Y(Y)j个)是DV-Curve的点。(X(X)0,Y(Y)0)=(0,0)是起点(X(X)结束,Y(Y)结束)是终点。
在这个模型中,每个S公司我给出了。我们可以计算每个点(X(X)j个,Y(Y)j个)然后用直线连接所有点。这样,我们就可以得到DV-Curve。
在这个模型中,每个点(X(X)j个,Y(Y)j个)给出了DV-曲线。我们可以计算每个S公司我根据这个等式,然后将每个S公司我逐一地。所以我们可以获得DNA序列序列=S公司1S公司2…S公司我…S公司n个.
2.3 DV-Curve的高级特性
在本小节中,我们使用数学方法和实验结果证明了DV-Curve的一些高级特性,如下所示:
还原为荒谬
假设DV-Curve中有一个或多个电路。因此,必须存在两个相互重叠的点。也就是说,我≠j个必须存在,使(X(X)我,Y(Y)我)=(X(X)j个,Y(Y)j个). 所以X(X)我=X(X)j个.根据方程式(三)和(4),X(X)我=我和X(X)j个=j个.因此我=j个。这与我≠j个因此,DV-Curve中没有电路和简并。 ▪
DNA序列和DV-Curves之间的对应是一对一的,没有信息丢失.
首先,我们证明了对于给定的DV-Curve,相应地存在唯一的DNA序列。
还原为荒谬
假设对应于一个DV-Curve,两个不同的DNA序列,序列1 =S公司11S公司12…S公司1我…以及序列2=S公司21S公司22…S公司2我…,存在。所以a米必须存在,使S公司1米≠S公司2米.自(X(X)10,Y(Y)10)=(X(X)20,Y(Y)20)=(0,0),根据方程式(1)和(2),一个k个必须存在,使Y(Y)12k个−1≠Y(Y)22k个−1或Y(Y)12k个≠Y(Y)22k个和0<k个≤米。这意味着有两个不同的DV-Curve。这与给定的DV-Curve相矛盾。
现在我们证明,对于给定的DNA序列,相应地存在唯一的DV-Curve。
还原为荒谬
假设对应于一个DNA序列,存在两条不同的DV-Curve,即Curves 1和Curves 2。所以有一点P(P)1(X(X)1米,Y(Y)1米)曲线1中必须存在与点不同的P(P)2(X(X)2米,Y(Y)2米)在曲线2中。也就是说米必须存在,使(X(X)1米,Y(Y)1米) ≠ (X(X)2米,Y(Y)2米). 根据方程式(三)和(4),X(X)1米=米和X(X)2米=米。这意味着X(X)1米=X(X)2米。因此,它必须Y(Y)1米≠Y(Y)2米.自(X(X)10,Y(Y)10)=(X(X)20,Y(Y)20)=(0,0),a我必须存在,使Y(Y)1我−Y(Y)1我−1≠Y(Y)2我−Y(Y)2我−1和0<我≤米根据方程式(5),一个k个必须存在,使S公司1k个≠S公司2k个这意味着必须有两个不同的DNA序列。这与一个给定的DNA序列相矛盾。
因此,DNA序列和DV-Curves之间的对应是一对一的,没有信息丢失。 ▪
终点的X坐标值表示DNA序列的长度。这是n=X(X)结束/2.
根据方程式(4),X(X)结束= 2n个.所以n个=X(X)结束/2 ▪
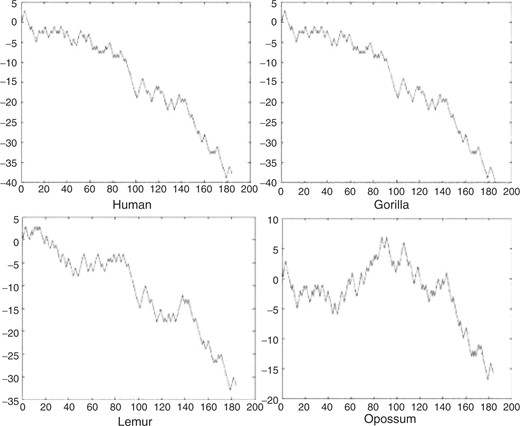
如所示图2b、 当DNA序列较短时,DV曲线非常清晰。此外,当DNA序列较长时,DV曲线也非常清晰。发件人图3,观察者很容易确定人类和负鼠之间的DNA序列差异很大,而从图1.
图3。
人β-珠蛋白基因全编码序列的DV-Curves(一)和负鼠(b条). 显然,它们非常清楚。观察者很容易识别出人类和负鼠之间的DNA序列差异很大。
通过检查绘图,人们可以立即掌握DNA碱基组成的信息.
如果有人想知道我-DNA序列中的第个字母是,他可以立即找到其值所在的点X(X)轴为2(我−1),然后他可以通过检查接下来的两点来了解这一点。
3 DV-曲线的应用
在本节中,给出了DV-Curve的三个应用。第一个是突变分析。我们可以通过检测相关的DV-Curves来定位突变并立即查明DNA中发生了什么。其他两个应用程序都是相似性分析。生物序列的相似性分析是生物信息学中最重要的部分之一,它可以分为两大类:序列比对和序列描述符比较。在这里,我们基于DV-Curve的数字特征提出了一个序列描述符比较。基于人脑在识别图形方面比计算机强大得多的事实,我们还提出了一种通过检测DV曲线进行相似性分析的方法。生物学家可以根据自己的需要选择方法。最后,介绍了一种可以很容易地绘制DV曲线的软件。
3.1基于DV-曲线目视检查的突变分析
由于物种的形态和生理特征最终由DNA携带的遗传信息控制,这些特征的突变变化是由于DNA分子的一些变化。因此,定位突变并找出DNA中发生了什么非常重要。核苷酸的这些变化可分为三种基本类型。它们是一个核苷酸替换另一个核苷酸(包括由嘧啶对嘧啶和嘌呤对尿嘧啶组成的转换,以及由嘧啶对尿嘧啶和嘌嘧啶对嘧啶组成的颠倒)、插入和删除。
传统的突变定位方法是基于计算机搜索技术的。例如,黄等。(2008)设计了一种快速定位突变的算法。然而,如果我们在一个图中绘制序列,我们可以立即定位突变。该方法的细节将在下一段中介绍。
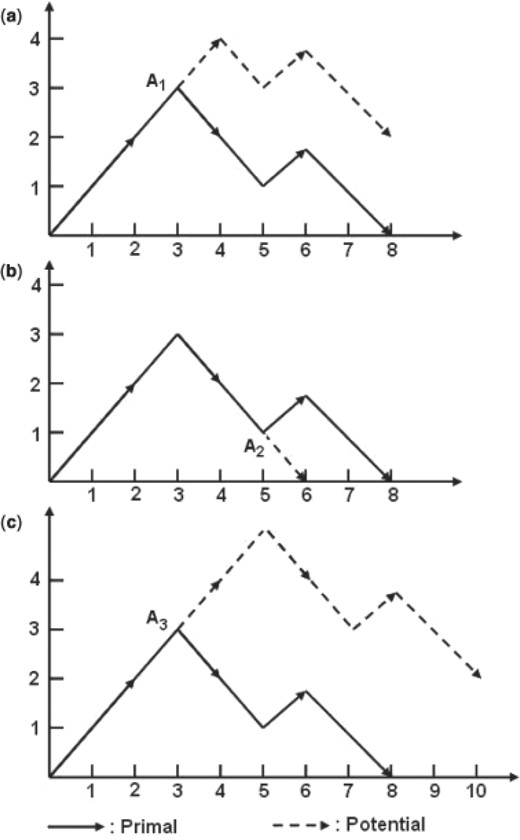
我们在同一个图中分别用黑线和红线绘制原始序列和势序列。显然,如果两条DV-Curves完全重叠,则潜在序列中不会发生突变。如果两个DV-Curves的某些部分不同,则必须发生一个或多个突变。因为DV-Curve沿X(X)轴代表每个核苷酸,无论是A、T、C还是G,突变都可以通过末端重叠点表示:哪里k个是DNA序列中突变的位置,j个是结束重叠点的值X(X)轴。例如,如所示图4a中,A类1是结束重叠点。根据方程式(6),j个= 3,k个=2,第二个碱基发生突变。在知道突变的位置后,我们可以根据方程式找出DNA中发生了什么(5). 例如,如所示图4a、 在第二个碱基的原始序列中有一个T,而在这个位置的电位序列中有个a。然后,通过比较曲线的端点,我们可以知道突变属于哪种类型,因为曲线的端点可以根据属性2.3指示序列的长度。例如,图4a属于替换,因为它们的长度相同;图4b属于缺失,因为潜在序列比原始序列短;图4c属于插入,因为潜在序列比原始序列长。
图4。
基于DV-Curves目视检查的突变分析。观察员很容易分类(一)作为替代(b条)作为删除和(c(c))作为插入。观察者也可以立即定位突变。
3.2基于序列描述符比较的相似性分析
序列描述符比较是进行相似性分析的主要方法之一。它基于DNA序列的定量表征,由序列派生的有序描述符集,例如各种矩阵的归一化特征值。例如,Randić等。(2003年a)提议E类矩阵,M(M)/M(M)矩阵,L(左)/L(左)矩阵和L(左)k个/L(左)k个矩阵,然后使用其特征值作为Randić中的描述符等。(2003年3月). 这些方法被证明是有用的,并被许多作者使用。然而,当DNA序列很长时,这些矩阵变得太大,无法计算特征值。
在我们的工作中,我们使用24分量向量作为描述符来对DNA序列进行数字表征,然后基于这些描述符进行相似性分析。
对于DV-Curve,我们可以计算厘米xy公司(廖等。,2005)具体如下: 对于一个序列,我们可以得到4!=通过将A、T、C、G分配给4中的基本双向量,可以得到24条不同的DV-Curve!方式。例如,我们可以指定四个核苷酸,如图2a.我们还可以将其分配如下: 所以我们可以得到描述符Vector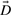 如下:
如下: 基本假设是,如果两个载体指向相似的方向,则由24组分载体表示的两个DNA序列相似。假设有两个物种我和j个,描述符向量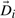 和
和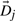 这些向量之间的相似性可以通过计算向量端点之间的欧氏距离来计算。距离d日ij公司在这两个向量之间是
这些向量之间的相似性可以通过计算向量端点之间的欧氏距离来计算。距离d日ij公司在这两个向量之间是 欧氏距离越小,DNA序列越相似。也就是说,进化密切相关物种之间的距离较小,而进化不同物种之间的差距较大。
该方法在11个物种的β-珠蛋白基因第一外显子的编码序列上进行了说明。相似度矩阵如所示表1.观察表1,我们发现人类-大猩猩和山羊-牛是最相似的,而负鼠(和Gallus)往往与其他动物更不同。
表1。基于描述性向量端点间欧氏距离的11个物种β-珠蛋白基因第一外显子编码序列的相似矩阵(1.0e+3)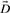
| 物种. | 人类. | 山羊. | 负鼠. | 盖洛斯. | 狐猴. | 鼠标. | 兔子. | 老鼠. | 大猩猩. | 牛. | 黑猩猩. |
|---|
| 人类 | 0 | 0.4769 | 1.1863 | 1.1559 | 0.4998 | 0.4440 | 0.5349 | 0.5270 | 0.2633 | 0.3606 | 0.9572 |
| 山羊 | | 0 | 1.2662 | 0.9361 | 0.6349 | 0.8413 | 0.3555 | 0.5968 | 0.3438 | 0.2121 | 0.9230 |
| 负鼠 | | | 0 | 1.3138 | 1.2384 | 0.9605 | 1.4799 | 0.9517 | 1.3756 | 1.2054 | 2.1057 |
| 加卢斯 | | | | 0 | 1.4916 | 1.2180 | 1.2854 | 1.3206 | 1.1030 | 1.1007 | 1.6580 |
| 狐猴 | | | | | 0 | 0.7792 | 0.4797 | 0.3045 | 0.5905 | 0.4249 | 1.0415 |
| 鼠标 | | | | | | 0 | 0.9663 | 0.6989 | 0.6872 | 0.7414 | 1.3649 |
| 兔子 | | | | | | | 0 | 0.6105 | 0.3822 | 0.2862 | 0.6999 |
| 老鼠 | | | | | | | | 0 | 0.6614 | 0.4172 | 1.2587 |
| 大猩猩 | | | | | | | | | 0 | 0.3139 | 0.7465 |
| 牛 | | | | | | | | | | 0 | 0.9253 |
| 黑猩猩 | | | | | | | | | | | 0 |
| 物种. | 人类. | 山羊. | 负鼠. | 加卢斯. | 狐猴. | 鼠标. | 兔子. | 老鼠. | 大猩猩. | 牛. | 黑猩猩. |
|---|
| 人类 | 0 | 0.4769 | 1.1863 | 1.1559 | 0.4998 | 0.4440 | 0.5349 | 0.5270 | 0.2633 | 0.3606 | 0.9572 |
| 山羊 | | 0 | 1.2662 | 0.9361 | 0.6349 | 0.8413 | 0.3555 | 0.5968 | 0.3438 | 0.2121 | 0.9230 |
| 负鼠 | | | 0 | 1.3138 | 1.2384 | 0.9605 | 1.4799 | 0.9517 | 1.3756 | 1.2054 | 2.1057 |
| 盖洛斯 | | | | 0 | 1.4916 | 1.2180 | 1.2854 | 1.3206 | 1.1030 | 1.1007 | 1.6580 |
| 狐猴 | | | | | 0 | 0.7792 | 0.4797 | 0.3045 | 0.5905 | 0.4249 | 1.0415 |
| 鼠标 | | | | | | 0 | 0.9663 | 0.6989 | 0.6872 | 0.7414 | 1.3649 |
| 兔子 | | | | | | | 0 | 0.6105 | 0.3822 | 0.2862 | 0.6999 |
| 老鼠 | | | | | | | | 0 | 0.6614 | 0.4172 | 1.2587 |
| 大猩猩 | | | | | | | | | 0 | 0.3139 | 0.7465 |
| 牛 | | | | | | | | | | 0 | 0.9253 |
| 黑猩猩 | | | | | | | | | | | 0 |
表1。基于描述向量端点间欧氏距离的11个物种β-珠蛋白基因第一外显子编码序列的相似矩阵(1.0e+3)
| 物种. | 人类. | 山羊. | 负鼠. | 加卢斯. | 狐猴. | 鼠标. | 兔子. | 老鼠. | 大猩猩. | 牛. | 黑猩猩. |
|---|
| 人类 | 0 | 0.4769 | 1.1863 | 1.1559 | 0.4998 | 0.4440 | 0.5349 | 0.5270 | 0.2633 | 0.3606 | 0.9572 |
| 山羊 | | 0 | 1.2662 | 0.9361 | 0.6349 | 0.8413 | 0.3555 | 0.5968 | 0.3438 | 0.2121 | 0.9230 |
| 负鼠 | | | 0 | 1.3138 | 1.2384 | 0.9605 | 1.4799 | 0.9517 | 1.3756 | 1.2054 | 2.1057 |
| 加卢斯 | | | | 0 | 1.4916 | 1.2180 | 1.2854 | 1.3206 | 1.1030 | 1.1007 | 1.6580 |
| 狐猴 | | | | | 0 | 0.7792 | 0.4797 | 0.3045 | 0.5905 | 0.4249 | 1.0415 |
| 鼠标 | | | | | | 0 | 0.9663 | 0.6989 | 0.6872 | 0.7414 | 1.3649 |
| 兔子 | | | | | | | 0 | 0.6105 | 0.3822 | 0.2862 | 0.6999 |
| 老鼠 | | | | | | | | 0 | 0.6614 | 0.4172 | 1.2587 |
| 大猩猩 | | | | | | | | | 0 | 0.3139 | 0.7465 |
| 牛 | | | | | | | | | | 0 | 0.9253 |
| 黑猩猩 | | | | | | | | | | | 0 |
| 物种. | 人类. | 山羊. | 负鼠. | 加卢斯. | 狐猴. | 鼠标. | 兔子. | 老鼠. | 大猩猩. | 牛. | 黑猩猩. |
|---|
| 人类 | 0 | 0.4769 | 1.1863 | 1.1559 | 0.4998 | 0.4440 | 0.5349 | 0.5270 | 0.2633 | 0.3606 | 0.9572 |
| 山羊 | | 0 | 1.2662 | 0.9361 | 0.6349 | 0.8413 | 0.3555 | 0.5968 | 0.3438 | 0.2121 | 0.9230 |
| 负鼠 | | | 0 | 1.3138 | 1.2384 | 0.9605 | 1.4799 | 0.9517 | 1.3756 | 1.2054 | 2.1057 |
| 加卢斯 | | | | 0 | 1.4916 | 1.2180 | 1.2854 | 1.3206 | 1.1030 | 1.1007 | 1.6580 |
| 狐猴 | | | | | 0 | 0.7792 | 0.4797 | 0.3045 | 0.5905 | 0.4249 | 1.0415 |
| 鼠标 | | | | | | 0 | 0.9663 | 0.6989 | 0.6872 | 0.7414 | 1.3649 |
| 兔子 | | | | | | | 0 | 0.6105 | 0.3822 | 0.2862 | 0.6999 |
| 老鼠 | | | | | | | | 0 | 0.6614 | 0.4172 | 1.2587 |
| 大猩猩 | | | | | | | | | 0 | 0.3139 | 0.7465 |
| 牛 | | | | | | | | | | 0 | 0.9253 |
| 黑猩猩 | | | | | | | | | | | 0 |
3.3基于DV-曲线目视检查的相似性分析
自1981年Smith和Waterman开发出动态规划算法以来,已有大量的比对算法来识别两个DNA序列是否相似。这些方法很有效。然而,由于运行时间太长,MSA(多序列比对)的效率会降低。此外,王和江证明MSA是一个NP完全问题(1990). 到目前为止,大多数专家认为不可能构建一个确定性多项式算法来处理NP完全问题。这意味着,使用当今任何可用的计算能力,解决即使是中等规模的NP完全问题所需的时间也很容易达到数十亿或数万亿年。
众所周知,人脑在识别数字方面比计算机强大得多。我们大脑的这种优势对我们在多个序列中进行相似性分析非常有帮助。因此,开发一种简单、清晰、独特、2D和非退化的DNA序列图形表示是可取的。分子生物学家可以使用这种图形表示作为一种直观的工具,从许多不同的DNA序列中找出与目标序列最相似的序列,然后使用对齐算法进行确认。
DV-Curve可以非常方便地用于完成这项工作。如图所示图5,这四个物种的DV曲线的所有趋势都有些相似,并且下降得越来越快。从DV-Curve的构建可以看出,G的含量远远大于A的含量,因此这四个序列在某种程度上是相似的,观察者也可以很快发现,人类和大猩猩的DNA序列在这四个物种中是最相似的,因为它们有最相似的DV-Curves图。这一过程的运行成本非常低,这归功于强大的人脑。
图5。
人类、大猩猩、狐猴和负鼠β-珠蛋白基因第一编码序列的DV-曲线。观察者很容易判断大猩猩与人类最相似。
3.4软件
为了方便生物学家,开发了一个非常容易使用的软件。输入DNA名称及其DNA序列,然后单击“绘制”。将立即创建相应的DV-Curve。如果要在同一窗口中绘制多条DV-Curve,可以重复上述步骤。
4结论
DV-Curve提供了一种直接绘制DNA序列的方法,无退化和信息丢失,但无论序列是否长,在二维空间都具有良好的可视化效果。它还可以反映DNA序列的长度,并可用于DNA序列的突变分析和两类相似性分析。相应的软件已经开发出来,将对生物学家有所帮助。
基金国家自然科学基金项目(6067410630870826、60703047、60533010);新世纪高校优秀人才计划(NCET-05-0612);教育部博士项目基金(20060487014);武汉市晨光项目(200750731262);HUST-SRF(2007Z015A);湖北省自然科学基金(2008CDB113和2008CDB180)。
利益冲突:未声明。
参考文献
, . DNA序列的新型4D数值表示
, 化学。物理。莱特。
, 2005
,卷。 407
(第63
-67
) 等低简并度DNA序列的一种新的二维图形表示
, 化学。物理。莱特。
, 2001
,卷。 350
(第106
-112
) . 用H曲线法对长DNA序列进行图形表示——当前结果和未来展望
, 生物技术
, 1989
,体积。 7
(第710
-720
) , . H曲线,一种新的核苷酸序列表示方法,特别适用于长DNA序列
, 生物学杂志。化学。
, 1983
,体积。 258
(第1318
-1327
) 等H-L曲线:DNA序列的新型二维图形表示
, 化学。物理。莱特。
, 2008
,卷。 462
(第129
-132
) . 基因结构的混沌博弈表示
, 核酸研究。
, 1990
,卷。 18
(第2163
-2170
) , . 基于核苷酸碱基非重叠三联体的DNA序列相似性/差异性分析
, 化学杂志。Inf.计算。科学。
, 2004
,卷。 44
(第1666
-1670
) 等DNA序列二维图形表示的应用
, 化学。物理。莱特。
, 2005
,卷。 414
(第296
-300
) . DNA序列结构的一种新的图形表示和分析。一: 珠蛋白基因的方法学和应用
, 货币。科学。
, 1994
,卷。 66
(第309
-314
) , . PNcurve:DNA序列的三维图形表示及其数值表征
, 化学。物理。莱特。
, 2007
,卷。 442
(第434
-440
) , . 基于双核苷酸的DNA序列二维图形表示
, 化学。物理。莱特。
, 2007
,卷。 440
(第139
-144
) 等一种新的基于双核苷酸的DNA序列三维图形表示
, J.西奥。生物。
, 2007
,卷。 249
(第681
-690
) 等DNA序列的新型二维图形表示及其数值表征
, 化学。物理。莱特。
, 2003
,卷。 368
(第1
-6
) 等基于新型二维图形表示的DNA序列相似性/差异性分析
, 化学。物理。莱特。
, 2003
,卷。 371
(第202
-207
) , . 常见分子子序列的识别
, 分子生物学杂志。
, 1981
,卷。 147
(第195
-197
) , . 关于多序列比对的复杂性
, J.计算。生物。
, 1994
,卷。 1
(第337
-348
) 等DB-Curve:一种新的DNA序列二维可视化和表示方法
, 化学。物理。莱特。
, 2003
,卷。 367
(第170
-176
) , . 一类新的DNA序列二维图形表示及其应用
, 化学。物理。莱特。
, 2004
,卷。 398
(第318
-323
) , . Z曲线,可视化和分析DNA序列的直观工具
, 生物分子杂志。结构。动态。
, 1994
,卷。 11
(第767
-782
) 等Z曲线数据库:基因组序列的图形表示
, 生物信息学
, 2003
,卷。 19
(第593
-599
)
作者注释
©作者2009。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oxfordjournals/org






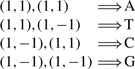
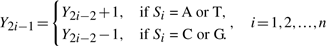
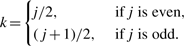
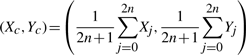
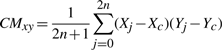
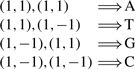
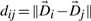


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}