摘要
1简介
尽管有许多工具可用于高通量微阵列数据处理(绅士等人。,2004; 赛义德等人。,2003)基于LC-MS的定量自下而上蛋白质组学测量数据(即无标签方法、稳定同位素标记方法、光谱计数方法以及精确质量和时间标记方法)与这些工具设计解决的问题不同。与蛋白质组学数据相关的主要问题之一通常是缺失值的程度,这主要是由于接近检测阈值的物种数量较多,导致数据集不平衡。此外,蛋白质组学数据涉及另一个级别的分组或“汇总”信息,以将肽映射到蛋白质。肽丰度通常用于推断相应的蛋白质丰度。
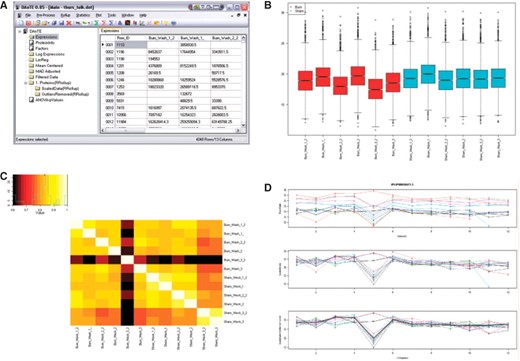
为了解决蛋白质组学数据的常见问题,数据分析工具扩展(DanTE)易于扩展。虽然目标应用是高通量蛋白质组学,但DAnTE也已成功用于微阵列数据分析,并且可以很容易地应用于具有类似特征的其他形式的高通量“组学”数据(例如代谢组学数据)。DAnTE用户界面的屏幕截图如所示图1.
图1。
DAnTE的代表性截图。(A类)左侧的数据网格和导航面板;(B类)对数转换数据的箱线图;(C类)显示可能的异常数据集的一组数据的相关性热图;(D类)肽对蛋白汇总来自RRollup方法(从上到下的面板:原始数据;缩放数据;去除离群值后,中间曲线显示为粗黑线)。
2说明
2.1依赖性
DAnTE的图形用户界面(GUI)使用C#语言实现,核心算法在开源R统计环境中实现(R开发核心团队,2008). DAnTE在.NET 2.0框架内的Microsoft WindowsXP平台上运行。R和C#/之间的连接。NET环境是通过使用开源R(D)COM服务器应用程序(Baier和Neuwirth,2007). 这种独特的环境选择使DAnTE成为一种非常用户友好的软件工具,尽管它无法集成到流行的Bioconductor中(绅士等人。,2004)项目。
2.2应用特点
2.2.1数据加载
DAnTE的输入数据可以是存储表格数据的任何文件,包括平面文件(CSV或tab分隔的文本文件)和Microsoft Excel文件。数据加载机制的一个独特特征是,它保留了肽对蛋白映射信息,供以后绘制属于特定蛋白质的肽以及肽对蛋白汇总方法中使用。此外,DAnTE还可以处理SEQUEST(工程等人。,1994)结果并创建光谱计数表。
2.2.2因子定义
在实验设计中,因子用于捕捉固定和随机效应。例如,生物条件是一个固定的影响因素,而用于分离样品的液相色谱(LC)柱列表可以被视为随机效应。这些信息对DAnTE的归一化、插补和假设检验方法至关重要。可以在加载数据后声明因子,也可以从平面文件加载因子。
2.2.3调查地块
DAnTE中可以绘制各种统计图,包括直方图、方框图、相关图和MA(或R-I:比率强度)图。这些曲线图有助于用户评估研究数据集内的再现性,并选出有问题的数据集,以便将其排除在进一步分析之外。
2.2.4数据规范化
由于标准化可以说是下游数据分析中最重要的一步,DAnTE采用了几种标准化方法,这些方法已经成功地测试了两种蛋白质组数据(Callister等人。,2006)和微阵列基因组数据(Quackenbush,2002; 斯迈思等人。,2003). 其中包括稳健线性回归方法、洛斯方法和分位数归一化方法。此外,还可以使用基于中值绝对偏差(MAD)和中心趋势调整方法的全球强度调整。
2.2.5缺失值插补
在高通量蛋白质组学中,由于缺失值而导致的数据集不完整是常见的。由于输入这些值是一个备受争议的话题(Troyanskaya等人。,2001)DAnTE提供了几种简单的方法,以及一些可供选择的高级算法。简单的方法允许用户使用数据集平均值/中值或预先选择的常量填充缺失的值。高级方法包括基于用户定义因子的行平均值填充、K最近邻插补(KNNimpute)和基于奇异值分解的插补(SVDimpute)。
2.2.6肽-蛋白质汇总
在大多数蛋白质组学方法中,肽测量值被汇总到相应的蛋白质丰度。理想情况下,来自单个蛋白质的所有肽应具有类似的丰度,表现为类似的信号强度;然而,在现实中,许多因素,如消化效率、电喷雾电离效率等,都会影响肽的鉴定和丰度或信号强度。在DAnTE中可用的RRollup方法中,来源于同一蛋白质的肽首先根据选定的参考肽进行缩放,以使生物条件下的所有肽谱达到相同水平,然后取平均值以获得蛋白质丰度。在标度过程中,选择观察结果最多的肽作为参考肽,并将其在数据集中的总丰度用作决胜点。在ZRollup方法中,缩放方法类似于z(z)-首先对来源于单个蛋白质的肽进行评分(除了使用中位数而不是生物条件下肽谱的平均值),然后对缩放后的肽进行平均,以获得相对蛋白质丰度。在RRollup和Zrollup方法中,使用Grubb的离群值检验(Grubbs,1969). 在第三种QRollup方法中,根据用户选择的丰度截止值选择肽,并将蛋白质丰度计算为这些选定肽的平均值。
2.2.7分析算法
DAnTE提供了几个特征鲜明的算法来进一步探索数据中的模式。传统的主成分分析(Jolliffe,2002)并且相关的分数和负荷图可以作为一种无监督的方法来发现数据中的主要变化。相反,偏最小二乘法(Wold等人。,1984)DAnTE中的可用信息可用作判别程序,其中分组信息是使用因子分配的。层次结构和k个-means在特征/样本上的聚类方法也可用作热图绘制功能的一部分。
2.2.8假设检验
使用边际平方和(Fox,1997)和混合车型(Pinheiro和Bates,2000)包含在DAnTE中。用户还可以在多元方差分析(ANOVA)中测试因素之间的交互作用。这个q个-值也与第页-值以控制多次测试中的错误发现率(Storey,2002). 此外,DAnTE可以通过使用Shapiro–Wilks检验来检查数据是否符合正态分布,并在正态假设不成立时采用两种非参数假设检验(Wilcoxon秩和检验和Kruskal–Wallis检验)。
3总结
DAnTE是一个完整的下游分析工具,它集成了大量用于大规模自下而上蛋白质组学数据的算法。该工具具有交互式GUI界面,并利用R统计环境的强大功能;它的独特之处在于它能够处理不完整的数据,并将肽合成蛋白质。尽管DAnTE是专门用于分析蛋白质组学数据的,但它在基因组微阵列数据上表现同样出色。
致谢
作者感谢Joel Pounds、Susan Varnum和Kim Hixson的许多建议和广泛测试;以及Thomas O.Metz为早期方法开发提供数据和支持。
基金:本研究的一部分得到了国家普通医学科学研究所(NIGMS,大规模合作研究拨款U54 GM-62119-02)、NIH国家研究资源中心(RR18522)、太平洋西北国家实验室(PNNL)实验室指导研究与开发(LDRD)计划(W.-J.Q.)的支持和国家过敏症和传染病研究所NIH/DHHS(通过机构间协议Y1-AI-4894-01)。工作在PNNL的环境分子科学实验室进行,该实验室是由美国能源部(DOE)生物和环境研究办公室赞助的国家科学用户设施。PNNL由巴特尔根据合同DE-AC05-76RLO-1830为DOE运营。
利益冲突:未声明。
参考文献
, . , 2007
(上次访问日期2008年5月23日) 等消除与质谱和无标记蛋白质组学相关的系统偏差的标准化方法
, 蛋白质组研究杂志。
, 2006
,卷。 5
(第277
-286
) 等蛋白质数据库中肽串联质谱数据与氨基酸序列的关联方法
, 《美国社会质谱学杂志》。
, 1994
,卷。 5
(第976
-989
) . 应用回归分析、线性模型和相关方法
, 1997
加利福尼亚州千橡树
Sage出版物
等生物导体:计算生物学和生物信息学的开放软件开发
, 基因组生物学。
, 2004
,卷。 5
第页。 80兰特
. 检测样品中异常观察值的程序
, 技术计量学
, 1969
,卷。 11
(第1
-21
) , . , S和S-PLUS中的混合效应模型
, 2000
纽约
施普林格
. 微阵列数据规范化和转换
, 自然遗传学。
, 2002
,卷。 32
(第496
-501
) R开发核心团队
R: 统计计算语言和环境
, 2008
奥地利维也纳
R统计计算基金会
等TM4:用于微阵列数据管理和分析的免费开源系统
, 生物技术
, 2003
,体积。 34
(第374
-378
) 等cDNA微阵列数据分析中的统计问题
, 方法分子生物学。
, 2003
,卷。 224
(第111
-136
) . 错误发现率的直接方法
, J.R.统计社会服务。B Stat.方法。
, 2002
,卷。 64
(第479
-498
) 等DNA微阵列的缺失值估计方法
, 生物信息学
, 2001
,卷。 17
(第520
-525
) 等通过主成分和pls-类模式和定量预测关系对数据表进行建模
, 阿拉鲁西斯
, 1984
,卷。 12
(第477
-485
)
作者笔记
©作者2008。牛津大学出版社出版。保留所有权利。有关权限,请发送电子邮件至:journals.permissions@oxfordjournals/org








{kind=link}