摘要
缩写名称“mfold web server”描述了万维网(WWW)上可用于预测单链核酸二级结构的许多密切相关的软件应用程序。此web服务器的目标是为广大科学界提供对RNA和DNA折叠和杂交软件的轻松访问。通过使用通用的web GUI(图形用户界面),服务器绕过了此软件的可移植性问题。详细输出的形式包括有或无可靠性信息的结构图、单链频率图和“能量点图”,可用于折叠单个序列。各种“批量”服务器提供的信息较少,但时间较短,一次最多可提供数百个序列。mfold web服务器的门户是http://www.bioinfo.rpi.edu/applications/mfold。此URL将被称为“MFOLDROOT”。
收稿日期:2003年2月14日;修订并接受2003年4月7日
简介
RNA二级结构的概念始于多蒂和弗雷斯科的工作(1,2). Tinoco及其同事开始使用最近邻能量参数通过能量最小化预测RNA二级结构(折叠)(三——6)还有Delisi和Crothers(7). 借鉴序列比对的动态编程方法,许多人独立开发了RNA二级结构预测的高效算法(8——13).
我自己的早期RNA折叠程序(12,14)计算了RNA序列的单个最小能量折叠。它们在20世纪80年代很受欢迎,修改后的版本被纳入了UWGCG(威斯康星大学遗传计算机组)的程序套件中(15);当这个团体离开威斯康星大学成立一家私人公司时,这个缩写被缩短为GCG。最初,他们使用Salser总结的自由能参数(16). 1986年后,使用了特纳小组(Doug H.Turner,纽约州罗切斯特大学化学系)的自由能(17).
RNA折叠的“mfold”软件开发于20世纪80年代末(18). “m”只是指“multiple”。核心算法预测最小自由能ΔG公司,以及必须包含任何特定碱基对的折叠的最小自由能。任何碱基对,第页我−第页j个,在我th核苷酸和j个折叠中包含的th核苷酸不超过δδG公司从最小值开始,绘制成三角形图,称为“能量点图”。碱基对第页我−第页j个按行打印我和列j个自由能增量δδG公司,已被选中先验的由用户选择“次优百分比”,P(P)由此,δδG公司计算为P(P)/100 |ΔG公司|. 这个自由能量增量内的碱基对是自动选择的,或者是由用户选择的,并且包含所选碱基对的折叠被计算出来。它们具有最小自由能,条件是包含所选择的碱基对。mfold包的描述和使用出现在许多文章中(19——22). 还描述了密切相关的“RNAstructure”程序(23,24).
特纳小组多年来发表了许多文章,详细介绍了RNA折叠参数的发展。这些文章的一个子集是我称之为“主要作品”的,总结了当前的技术状态。mfold包的版本1使用了Freier描述的自由能等。(17). 2.1至2.3版使用了Walter的参数等。(25)尽管尚未完成将同轴堆叠参数纳入最小化算法。当前版本3的软件使用来自马修斯的自由能数据等。(26).
mfold软件的DNA折叠预测始于1996年,当时通过与SantaLucia小组的合作将DNA特定参数添加到mfold包中(John SantaLuica Jr.,密歇根州底特律韦恩州立大学化学系)。圣卢西亚对这些数据进行了描述(27). DNA堆积(27),单个不匹配(28——32)和悬垂的末端(33)参数已在圣卢西亚实验室测量。其余终端堆叠和环路参数由圣卢西亚估算,并通过约翰·圣卢西亚的个人通信纳入mfold包。他们仍然没有发表。1999年,对[Na+],[Mg++]被合并到mfold包中(34).
mfold web服务器于1995年秋季在华盛顿大学医学院首次创建。1996年春季增加了DNA折叠参数。从1995年到2000年秋季,该服务器运行在SGI工作站或多处理器服务器上(Silicon Graphics,Inc.1600 Amphithetre Pkwy.,Mountain View,CA 94043),以及双处理器“Intel/Solaris”平台上(Intel Corp.的Pentium 2处理器,Sun Microsystems的Solaris操作系统)。服务器于2000年10月迁移到伦斯勒理工学院(RPI)。它在双处理器“Intel/Linux”平台上运行(Linux操作系统由Red Hat,Inc.开发和销售,1801 Varsity Drive,Raleigh,NC 27606)。自2002年7月以来,mfold web服务器一直在由36个双处理器“Intel/Linux”工作站组成的集群上运行,这些工作站由IBM捐赠给RPI-Wadsworth联合生物信息中心(IBM Research,P.O.Box 218,Yorktown Heights,NY 10598)。该设备被授予RPI和沃兹沃斯中心(PI:M.Zuker)的SUR(共享大学研究)拨款。
自mfold web服务器问世以来,其使用量稳步增长。“quikfold”服务器是在我还在华盛顿大学时添加的。将要描述的其余服务器都是在过去两年内添加的。这些服务器已被大学、医学院、非营利组织、美国政府和军事实验室的研究人员以及世界各地的公司广泛使用。此外,服务器正在用于计算生物学方法的教学。
服务器内容和组织
mfold网络服务器包括许多单独的应用程序,用于预测核酸折叠、杂交和熔化温度(T型米s) ●●●●。
基本“mfold”服务器:输入
mfold web服务器上的原始应用程序处理每个提交(作业)折叠单个RNA或DNA序列。RNA和DNA的提交表格仅出于历史原因而分开。首先是RNA折叠。这些应用程序可以通过主门户或入口页面的“RNA折叠”或“DNA折叠”超链接进行访问。门户的URL可能会保持稳定,而单独应用程序的URL则会更改。默认的RNA折叠形式目前使用最新版本3.0的自由能(26). 这些建议用于大多数RNA折叠。然而,有一个链接从这个页面到什么是所谓的“RNA mfold版本2.3服务器”。该服务器使用旧的2.3版能量参数提供RNA折叠(25). 为什么使用较旧且不太准确的参数?原因是我们有这些旧参数的焓。与自由能参数一样,在37°C下测量焓。然而,假设它们在可能发生的温度范围内保持不变体内或者在实验室里。这使服务器能够将自由能外推到其他温度,并在这些温度下折叠。
序列名称。
可以在“输入序列名称:”文本字段中键入或粘贴(输入)序列名称。长名称被截断为40个字符。可以使用任何ASCII字符,包括八进制值大于200的字符 八进制(O)。“危险”字符“,<,>和',分别转换为',≪(253 O),Ş(273 O)和'(264 O)。字符\被删除。如果未输入名称,则序列名称将变为“作业ID”,其格式为yyMmmdd-hh-mm-ss,其中yy是年,Mmm是月,dd是日,hh是小时,mm是分钟,ss是秒。例如,2003年2月9日当地时间晚上8:23:06进入服务器的作业将被分配一个作业ID为03Feb09-20-23-06。如果在同一秒内有两个作业进入,则第二个作业会附加字母“a”,对于同一秒内的两个以上作业,依此类推。
顺序。
必须在序列文本区域框中输入序列。除“A–Z”和“A–Z”之外的所有字符都将被删除。小写字符转换为大写字符。对于RNA折叠,“T”或“T”转换为“U”,而“U”或“U”在DNA折叠中转换为“T”。例如,如果您输入

那么结果序列将是
而不是

字母“N”应用于未指定的基础。不允许配对。字母“B”、“D”、“H”和“V”分别表示“A”、“C”、“G”和“U/T”。这些核苷酸只有在其3′邻未配对时才能配对。本公约的目的是表示连接所示核苷酸及其3′邻体的磷酸二酯键的核酸酶裂解。当核酸酶消化数据指定敏感键时,它用于限制折叠。此外,字母“W”、“X”、“Y”和“Z”也分别表示“A”、“C”、“G”和“U/T”。这些核苷酸如果配对,应该只在螺旋末端配对。因此,mfold web服务器不支持IUPAC(国际纯粹和应用化学联合会)模糊的DNA字符约定(35)如表所示1.
约束。
约束框中的文本区域允许可选合并折叠约束。每个约束都由框中的一行组成,该行必须符合刚性格式。使用约束时,主结果页面上会出现一个标记为“序列注释说明”的超链接。
下面列出了各种类型的约束。允许以任何顺序使用任何形式的多个约束。
强制形成特定的碱基对或螺旋。命令
F i j k公司
将强制形成螺旋(如果k=1)
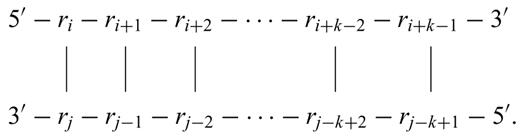
三人组(我,j个,k)指k连续的碱基对,其中第页我·第页j个是外部闭合碱基对。如果这些碱基对中的任何一个都不存在,那么将生成错误,作业将失败。通常的结果是一个输出页面,声明“Job aborted!”!没有结构!”。在输出页面的文本区域中,成功强制的碱基对用(“and”)下划线,而那些强制配对但无法配对的碱基对用“!”下划线。请注意,折叠代码不允许使用孤立的碱基对。那就是,即使第页我·第页j个是一个有效的基对,除非第页我+1·第页j个−1或第页我−1·第页j个+1是有效的基对。
禁止形成特定的碱基对或螺旋。命令
菲律宾
将禁止窗体的每一个碱基对第页i+小时·第页j−h, 0≤小时≤k,不会发生。在任何情况下都无法形成的禁止的碱基对将用“!”下划线在主输出页的文本区域中,但折叠将无误地进行。
强制一系列连续的碱基配对。命令
F i 0 k
(倒数第二个字符为“0”)将强制核苷酸第页我,第页我+1,第页我+2,…,第页我+k−1配对。当k=1.强制太多的碱基进行配对或强制标记为“N”的碱基配对将产生致命错误。
禁止字符串的连续碱基配对。该命令
P i 0千
(倒数第二个字符为“0”)将阻止核苷酸第页我,第页我+1,第页我+2,…,第页我+k−1配对。当k=1.强制过多的基成为单链可能会产生致命错误。
禁止一个由连续碱基组成的字符串与另一个字符串配对。该命令
菲律宾
会阻止核苷酸第页我,第页我+1,第页我+2,…,第页j个与核苷酸配对第页k,第页k+1,第页k+2,…,第页我(我≤j个和k≤我). 注意,如果我=k和j个=我,则该约束等价于禁止所有碱基对在内部细分市场第页我,…,第页j个.
其他折叠参数
RNA和DNA序列可以是线性的或圆形的。默认值为“线性”,但可以使用折叠表单上的选项按钮选择“圆形”。值得注意的是,折叠圆形核酸的算法比折叠线性核酸的算法简单。
使用3.0版能量规则,RNA折叠的折叠温度固定在37°C。对于2.3版参数的RNA折叠或DNA折叠,可以选择0到100°C之间的任何积分温度。
离子条件只能因DNA折叠而改变。对于RNA,离子条件固定在[Na+]=1 M和[Mg++]=0 对于折叠,这些相当于生理条件。以下约束适用:
[纳+]≥0.01 M、,
[镁++]≤0.1 M、 和
[纳+]≤0.3 M如果[Mg++]>0 M。
为了折叠,Na+可被视为等同于李+,K+和NH4+,而镁++相当于Ca++.
次优百分比,P(P),控制自由能增量δδG公司用于在能量点图中显示碱基对和计算次优折叠。自由能≤Δ的折叠中可能出现的碱基对G公司+δδG公司将绘制,并且只有自由能≤Δ的褶皱G公司+ δδG公司将进行计算。通常为δδG公司=P(P)/100|ΔG公司|,但四舍五入为1 kcal/mol或降至12 kcal/mol,如果超出此范围。
折叠数的上限是绝对限制。计算的折叠数可能小于此数量。通常情况下,它应该小于这个数字,因为通过正确选择P(P)和窗口参数。
窗口参数,W公司,控制计算的折叠数。它可以被认为是一个距离参数。2之间的距离 bp,第页我·第页j个和第页i′·第页j′可以定义为max{|我−我′|, |j个−j个′|}. 如果kmfold已经预测了-1个褶皱k第个折叠必须至少有W公司至少有一个距离的碱基对W公司从第一个k−1个折叠。除非满足此标准,否则不会将新折叠添加到输出列表中AsW公司增加时,预测的折叠数减少。如果W公司未指定,则mfold默认根据序列长度选择值。
如果成对基之间的最大距离参数,M(M),然后指定任何基对,第页我·第页j个,在线性分子的折叠中必须满足j个−我≤M(M)在圆形分子中,条件变为min{j个−我,N个 +我−j个}≤M(M),其中N个是序列长度。
即时作业与批处理作业。
当用户等待时,可以进行最多包含800个碱基的折叠序列。这是默认设置。此时,即时工单的折叠结果将被删除24-26 h后提交。对于长度从801到6000个碱基的序列,必须从相应的选项按钮中选择批次选项。在这种情况下,用户应该输入有效的电子邮件地址,尽管电子邮件地址总是受欢迎的,因为它们可以识别用户。对于批处理作业,按“折叠RNA”或“折叠DNA”提交按钮会将用户带到一个通知页面,该页面指示结果的URL以及折叠完成时将向其发送通知的电子邮件地址。如果立即访问结果页面的链接,则目标页面将不完整。它将只包含序列和一些其他输入信息。折叠完成后,必须“刷新”或“重新加载”此页面才能看到结果。结果的URL在48–51期间仍然有效 h、 然后删除结果。最多可折叠10个 如有特殊要求,可提供400个底座。折叠6000个底座目前大约需要1.5 h.标记为“查看折叠结果”的超链接将允许用户查看服务器上的任何结果,前提是这些结果没有被删除,并且用户从生成折叠的同一台计算机查看。也就是说,用户的IP地址必须相同。
输出参数
默认的常规图像分辨率值为每英寸72×72像素2用于png和jpg图像。低、中、高值分别为60×60、110×110和200×200。此参数对PostScript输出文件以及可以从中派生(“distilled”)的pdf文件没有影响。这些文件可以按需放大,而不会降低质量。Jpg和png图像将始终以每平方英寸72×72像素显示。更改分辨率将更改整个图像的大小。图像尺寸如表所示2.
绘制结构格式图,显示单个基础,或者在没有基础的情况下以大纲模式绘制。轮廓选项适用于非常大的折叠分子的图像。默认选择“Automatic”(自动)将使底座以多达800的折叠显示 nt和其他绘制的轮廓。如果用户打算放大结构以显示可以看到单个碱基的部分,那么为大序列选择“碱基”选项可能有意义。
可以关闭能量点图中的网格线。
默认情况下,将根据序列的长度对基数进行编号。默认值如表所示三。用户可以自由更改此值,也可以通过选择频率零来完全关闭基数。
用户可以通过选择旋转角度来改变折叠分子的自动选择方向。正值对应逆时针方向。
Zuker和Jacobson描述了结构注释(36). 绘制结构中的基底可以用“p-num”值进行注释,该值表示基底在δδ内所有折叠中的配对方式数量G公司最小能量。低值表示“定义明确”的基础。特别是,值0或1表示一个基总是单链的,或者总是分别与一个唯一的伙伴配对。基址在计算的折叠中单链的次数称为“ss-count”数,也可以使用这些数字对结构图进行注释。颜色方案显示在MFOLDROOT/www-NAR03/doc/colors.html最后,对于RNA折叠,“高光”选项允许用户指定序列中要突出显示的区域。选定的基础以绿色绘制,而其余的基础以黑色绘制。如果结构格式为“Bases”,则基本字符将以彩色绘制。如果结构格式为“大纲”,则表示基础的彩色点将叠加在梯形轮廓图上。如果使用默认的“自动”选项,则如果序列大小>800,则使用彩色点。然而,如果序列大小≤800,则二者都使用彩色点和底。底座在适当颜色的圆点内以黑色或白色显示。
折叠结果
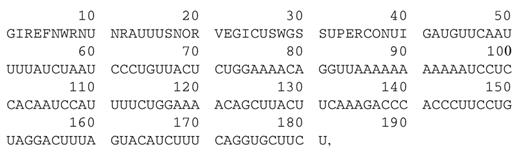
可通过以下来自初始结果页面的链接查看折叠结果。一些输入信息被复制在该页面的顶部的文本区域中。初始结果页面顶部的示例如图所示1.
能量点图。
能量点图以PostScript、png和jpg格式提供。“文本”超链接指向一个提供基本点图信息的纯文本文件。“istart”、“jstart”和“length”选项对应于(我,j个,k)定义螺旋的数字,如前所述。以十分之一kcal/mol为单位的整数中的“能量”。级别1对应于最佳折叠中的螺旋。2–4级对应于日益次优的折叠中的螺旋。从1到4的级别对应于点图中碱基对的默认颜色。PostScript超链接会导致能量点图的静态PostScripts图像。出租δG公司是包含一个碱基对的折叠的最小自由能,能量点图中的碱基对默认颜色(rgb Hex)如表所示4.
png和jpg超链接分别指向一个新页面,其中包含能量点图的png或jpg图像以及允许用户与点图交互或重新绘制点图的按钮。它被称为主点图页面。屏幕左上角将打开一个550×40(像素)的小“详细信息”窗口。选中“Click on a base pair to identify”单选按钮(默认设置)后,单击(左键单击)图像将导致(我,j个)所选碱基对的值及其δG公司值显示在“详细信息”窗口中。如果选择绘图的空白区域,δ的值G公司将被声明为“未定义”。
选择“单击图像并用下面的选项重画点图”按钮将激活下面的文本区域和五个选项按钮。单击点图中的点将创建新图像。如果放大系数选择>1,则新图像将围绕所选点进行放大;否则,将重新绘制整个点图。“能量增量(kcal/mole)”选项的值将是δ的上限G公司。例如,将其设置为零将只绘制最佳碱基对。点图中使用的不同颜色的数量可以更改。如果k是“Filter”选项的值,则只有大小至少为k将在重新绘制点图时绘制。“图像宽度”控制重画图像的分辨率(大小),如上所述。如果选择了PostScript“输出”选项,则会创建一个静态PostScripts图像。如果选择了png或jpg“输出”选项,则单击绘图将打开一个新窗口,其中包含重画的图像和与之前相同的一组控制按钮。这称为辅助点图窗口。从该二级窗口创建的新png或jpg图像将在二级窗口中绘制。类似地,从主点图窗口生成的新png或jpg图像将刷新辅助窗口。典型的用法是放大初始点图中的某个区域。然后可以精确地选择放大点图中的碱基对,使用户可以识别最佳或次最佳折叠中可能出现的特定螺旋。关闭辅助窗口不会影响其他窗口。关闭主窗口也将关闭“详细信息”窗口。
RNAML语法。
RNAML中也有计算的二级结构(37)格式。这种格式被提出作为交换RNA结构信息的语法。我们目前提供的输出与Universite®de Montre®al的Major集团维护的DTD(文档类型定义)文件兼容。此文件可在以下位置找到:http://www-lbit.iro.umontreal.ca/rnaml/current/rnaml.dtd.
一些额外的文件。
这些是包含可能对某些用户有用的信息的原始文本文件。如上所述,通过p-num值进行注释在服务器上可用。原始值也可用。h-num链接包含能量点图中每个螺旋的h-num值,从h-num的最低值到最高值进行排序。螺旋的h-num值是螺旋的“精细度”(38). h-num值较低的螺旋更容易被预测。运行的日志文件有时有助于查找作业失败的原因。
批量下载结果。
可以在单个压缩文件(.zip)或压缩tar文件(.tar.Z)中一次性下载所有计算出的文件夹。这些结构可以作为PostScript、png或jpg打印文件,也可以作为不同格式的原始文本文件。
这些文本文件对那些希望自己创建结构图的人很有用。一些文本格式之前已经介绍过(21,22). 有关详细说明,请访问MFOLDROOT/www-NAR03/doc/structure-format.html。重画结构或使用新的“sir_graph”程序创建的结构只能单独下载。
单股频率。
ss-count文件包含所有计算出的折叠中每个基的单链度的显式统计信息。第一行包含计算的折叠数。这个我接下来的第行包含我第个基数、基数本身以及它在所有计算出的折叠中的单链次数。“View plot”(查看绘图)按钮创建基数与ss-count的绘图。当“平均窗口”为米>1,然后为我th基数是米以为中心的底座我在地块末端附近可以使用较少的基底。放大倍数使用户能够放大感兴趣的区域。
结构输出。
“Structure i”的超链接指向包含我结构采用易于阅读的文本格式。此格式可能适用于查看小折页。它可以很容易地粘贴到文本文件中。如果序列非常大,并且结构图的分辨率很低,则查看文本文件的一部分可能比放大结构图的一部分更方便。
选择PostScript、.ct文件、RnaViz ct、Mac ct、GCG connect或XRNA-ss将生成指定格式的单个结构。在查看能量点图时,遵循png或jpg链接与遵循png和jpg连接类似。两者都会生成一个新页面,其中包含结构图的png或jpg图像以及允许用户与折叠进行交互或重新绘制折叠的按钮。它被称为主结构绘图页。屏幕左上角将打开一个550×150(像素)的“循环自由能量分解”小窗口。选中“单击一个碱基对以查看回路自由能量分解窗口中的相关结构细节”单选按钮(默认设置)后,单击(左键单击)图像将使所选碱基对的细节显示在小窗口中。窗口将显示循环的类型,包括由选定的基对关闭的堆栈。给出了该回路的自由能,并给出了基对的具体恒等式。如果所选点没有明确选择一个基对,则辅助窗口将打印一条消息,指示用户重试。
选择“单击图像并用下面的选项重画结构”按钮将激活下面的文本区域和四个选项按钮。单击结构图中的点将创建新图像。如果放大系数选择>1,则新图像将围绕所选点进行放大;否则将重新绘制整个结构。“annotation”(注释)按钮允许用户选择p-num或ss-count注释,即使最初没有选择这些注释。除非选择了注释选项,否则“注释类型”按钮无效。在这种情况下,将使用彩色基本字母、彩色点或包含基本字母的彩色点绘制带注释的结构,分别对应于选项“字符”、“点”或“两者”。“图像宽度”控制重画图像的分辨率(大小),如上所述。如果选择了PostScript“输出”选项,则会创建一个静态PostScripts图像。如果选择了png或jpg“输出”选项,则单击绘图将打开一个包含重画图像的新窗口。此页面上没有按钮。“Color Table”选项会生成一个表,该表显式显示结构图中使用的颜色与p-num或ss-count值之间的关系。p-num或ss-count文件超链接指向相应的原始文本文件。这称为二级结构窗口。从主点图窗口生成的新png或jpg图像将刷新次窗口。典型的用法可能是放大初始结构图中的某个区域,以便以合适的分辨率打印结构的一部分。它还可以帮助用户精确地选择碱基对。关闭辅助窗口不会影响其他窗口。关闭主窗口也将关闭“循环自由能量分解”窗口。
结构输出的新形式。
默认情况下绘制并可立即下载的结构图大部分由Bruccoleri和Heinrich的“naview”程序生成(39). “naview”程序创建独立于设备的ASCII绘图文件(后缀为plt2)。华盛顿大学为mfold软件包开发的软件从这些文件中创建PostScript、jpg和png图像。华盛顿大学创建了一个名为“sir_graph”的新绘图程序,用于显示核酸二级结构。它是一个在各种Unix平台上运行的交互式程序。非交互版本“sir_graph_ng”为mfold web服务器创建显示。主要结果页面上的“单击此处获取新的结构查看选项”超链接会导致“结构查看器”页面,该页面等效于上述主要窗口页面。此页面与通过“查看单个结构”部分中的“新”超链接可以访问的页面相同。
用户可以使用此新软件进行更广泛的选择。外环或外基对可以默认方式绘制,围绕圆排列,也可以绘制为“平面”,与之相交的所有螺旋线彼此平行。“Flat_Alt”选项以180°的角度绘制连续茎。背景可以是通常的白色或黑色。基本对符号可以是mfold web服务器常用的“点”,也可以是大多数二级结构显示器中常用的“线”。“Algorithm”可以是“Default”或“Simple”类型,这两种类型都可以避免词干重叠。“Default”和“Simple”算法以不同的方式绘制多分支循环。“自然”选项将每个循环画成一个完美的圆,茎以适当的角度伸出。这可能会为较小的折页产生令人满意的效果,但通常会为较大的折页带来令人绝望的混乱。与使用旧版naview程序显示结构一样,“Loop Free-Energy Decomposition”窗口将打开,允许用户识别精确的循环或堆栈,以及它们的自由能和闭合碱基对。此外,二级结构窗口页面可用于放大所选点。
热力学细节。
当温度无法改变时,“Thermodynamic Details”链接会指向html文件的一部分,其中包含特定折叠到循环和堆栈的整个分解,以及它们的自由能和闭合碱基对。碱基对的连续运行被概括为螺旋。当用户与折叠的png或jpg图交互时,此文件的小部分显示在“循环自由能量分解”窗口中。
当温度可能发生变化时,会出现一个更有吸引力的页面,给出自由能、焓、熵和估计值T型米.T型米使用简单的2状态模型进行计算。这假设分子要么如图所示折叠,要么完全单链。这种假设对于短分子来说是合理的。对于较大的分子,在预测值附近重新折叠可能有用T型米如果新的T型米“显著”变大,这表明给定的折叠可以重新排列成具有更高熔化温度的另一个折叠。服务器使用T型米=ΔH(H)/ΔS公司,式中ΔH(H)和ΔS公司分别是结构焓和熵。(由于熵的单位,实际公式为T型米=1000ΔH(H)/ΔS公司−273.15摄氏度)下面是常用的详细信息表。
结构点图。
“点图折叠比较”选项允许用户查看点图中计算出的折叠的任何子集。当计算两个或多个折叠时,此选项可用。用户可以通过选择相应的单选按钮来选择要显示的文件夹。默认情况是显示前两个折叠。为方便起见,提供了“全部”按钮。
所有折叠中出现的碱基对都是黑色的。那些出现在两个或两个以上,但不是所有的褶皱都是灰色的。否则,将根据结构为碱基对指定唯一的颜色。当计算超过15个结构时,该系统会崩溃。
在第一个按钮中选择png或jpg选项,然后按“进行比较”选项,将显示与一次能源点图页面非常相似的一次结构点图页面。选中默认的“Click on a base pair to identify”单选按钮,点击一个基对,将显示基对的标识,并列出包含该基对的计算结构。当选中“单击图像并用下面的选项重画点图”单选按钮时,单击点图将生成新的点图。如果选择png或jpg选项,则点图将位于“二级”结构点图窗口中。这与能量点图的二级点图窗口非常相似。放大率、输出格式和图像宽度参数是上述能量点图的常用参数。可以选择的“多色重叠”选项可以在多色模式下绘制其他灰色圆点,精确显示折叠中包含的碱基对。在mfold web服务器中,“点”实际上绘制为(通常是小的)正方形。在方形区域内绘制了不同颜色的梯形集合。它们指示哪些折叠包含该碱基对。点图必须以足够高的放大倍数绘制,以便能够看到此特征。当只计算了两个折叠时,就没有必要了。计算三个折叠时,结构1和2、1和3以及2和3中的灰色碱基对以三种不同的颜色绘制在左下角的三角形中,因此可以在不使用多色选项的情况下识别它们。
其他服务器
“quikfold”服务器:一次折叠多个序列。
许多用户对mfold web服务器上提供的复杂查看选项不感兴趣。有些可能满足于易于阅读的文本格式或“ct格式”文件,这些文件可以下载并在本地用于创建二级结构图。许多人希望在相同的条件下一次折叠多个序列。例如,用户可能希望预测数百个短分子信标的折叠和熔化温度。
因此,创建了“quikfold服务器”。它使用相同的“nafold2”程序在常规服务器中折叠序列,但使用不同的多分子模式。只允许直接作业,每个序列包含的核苷酸不超过600个。一次提交的序列数的理论上限为25 000,但我不建议提交那么多!上限取决于Internet、浏览器设置(如超时)和其他因素。例如,我确实知道,1000个长度为100的序列应该没有问题。
输入页面包含已经定义的按钮和文本区域。唯一的新项是序列输入框需要一个额外的字符。每个输入序列必须由(至少)一个分号“;”分隔。除“A–Z”和“A–Z”之外的所有其他字符都将在预处理步骤中删除。
quikfold服务器提供文本、ct、RNAML和热力学细节作为输出。在每种情况下,输出都是一个单独的、可能很大的文本文件。序列的命名方法是采用给定或默认的序列名,并在每个名称中添加“_i”,其中i的范围从1到序列数。折叠结果在40内被擦除 分钟到几个小时。无法折叠的单个序列不会中止整个作业。它将在输出中跳过。
“zipfold”服务器:仅预测最小自由能。
一些用户只需要序列的最小折叠能量。因此,创建了zipfold服务器。输入页面类似于quikfold服务器的页面。序列长度可达800。在一个例子中,一个用户提交了数千个作业;每个序列包含50个长度为500的随机序列。失败率小于1%。
由于速度很重要,并且请求的信息很少,因此对底层代码进行了简化,使其运行速度更快。在撰写本文时,在11个多一点的时间里处理了378个tRNA s.实际性能取决于服务器负载和难以控制的互联网相关因素。
底层代码已被剥夺了处理约束的能力。此外,“填充算法”被截断,因此只能计算最佳能量或单个最佳结构。新的实验版本也简化了序列输入。
所有输出都显示在一个主结果页面上。ΔG公司值排列在列表中。那些不能折叠的序列被赋予了巨大的正折叠能量。当前值为10 0
“T”米单链核酸服务器:双态熔化温度预测。
一些用户希望得到比最小折叠能量多一点的能量。他们还希望T型米在这种情况下T型米可以使用服务器。此服务器仅提供2.3版RNA折叠参数,因为估计需要焓T型米。输入与zipfold服务器相同。
这个T型米服务器使用与zipfold服务器相同的简化折叠程序,只是在这种情况下计算的是最小能量折叠。焓ΔH(H)然后使用适当的最近邻参数计算此折叠的。由此,很容易计算熵ΔS公司然后T型米,使用如上所述的2状态模型。
事实上,创建此服务器是为了“服务”Rouillard的“OligoArray 1”程序等。(40). OligoArray程序使用Java应用程序创建的直接请求,用单个DNA序列“命中”服务器。OligoArray的后续版本将放弃此低效过程。
杂交服务器:两股杂交。
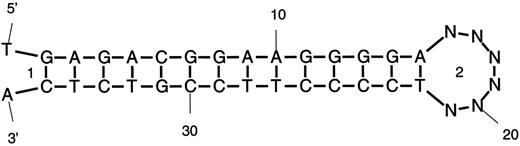
多年来,底层mfold软件已经添加了许多小改动,以适应“特殊请求”或应用程序。许多用户使用常规的mfold软件,无论是以“独立”的形式还是在网络上,来模拟两股RNA或DNA的杂交。他们采取了两个序列,A类=一1一2一三···一米和B类=b条1b条2b条三···b条米并创建单个序列,S公司=一1一2一三···一米n个1n个2···n个kb条1b条2b条三···b条米,通过串联A类具有B类使用一些非空字符,n个1···n个k作为链接器。
这种方法有两个问题。第一个不太严重的问题是k连接物残留物最终形成某种循环。如果A类和B类完美地混合,那么这个循环几乎肯定是发夹式循环。图中给出了这种折叠的示例2问题是错误的发夹自由能被施加到回路“2”。相反,它应该被视为外部循环,如“1”。此外,起始自由能Δ我,需要添加。这是4.1 37℃和1.96℃下RNA的kcal/mol 37°C时DNA的kcal/mol。
为了适应这种情况,mfold中添加了以下功能。当一个序列中出现三个连续的L(L代表链接器)时,它们被识别为链接器。如果它们出现在发夹形回路中,则该回路被视为外回路和Δ我也添加了。输出仍然包含连接体残基,但Δ的值G公司现在是正确的。
通过强制一个简单的杂交,连接体残基可能被迫处于发夹环中A类具有B类。这可以通过使用两个约束来实现
这些约束禁止所有分子内碱基对,因此只有允许的碱基对链接A类具有B类,除其他外,还迫使链接器残留物处于发夹式循环中。
然而,更严重的问题是T型米是胡说八道,因为总核酸浓度,C类两股杂交时必须考虑。正确的2状态估计T型米是哪里如果=2,如果A类≠B类和如果=4,如果A类=B类. 所有这些都是由混合服务器自动完成的。这两个序列,A类和B类,被输入到序列框文本区域,由分号“;”分隔。必须给出总核酸浓度。结果是一个包含作业IDΔ的简单输出页面G公司, ΔH(H), ΔS公司和T型米在一行中。没有给出单位,尽管它们被理解为kcal/mol、kcal/mole、cal/(K · 摩尔)和°C。
访问
mfold web服务器对所有用户开放。对商业用户没有任何限制。但是,用户应该知道服务器不安全,其他人可能会检测到流入和流出的数据。此外,查询信息无限期地存储在服务器上的日志文件中。这些日志文件被视为机密信息,尽管收集和传播了使用情况的总体统计数据。此外,只有当数据库搜索显示序列已经在公共数据库中时,才会选择一些提交的文档作为教学示例。
“GET”请求可能不会命中常规mfold服务器。只允许“POST”操作。所描述的其他应用程序也可能会受到“GET”请求的影响。直接使用Java应用程序攻击其他应用程序是允许的,并且将继续允许,直到服务器过载。这还没有发生。在华盛顿大学,每个工作日提交的折叠工作可能有100-200份,而在周末提交的则少得多。例如,2002年12月,我数了21个 688份提交给常规mfold web服务器,3128份提交给quikfold,35份 904到拉链,193 428至T型米和542到杂交服务器。
设备和组织
当前的web服务器运行在IBM Research于2001年秋季捐赠给RPI的设备上。表中给出了详细的硬件规格5.捐赠设备的总价值约为214美元 000美元。我们又捐款5000至10美元 000用于开关和定制设计的电源。所有设备最初都是在电子媒体学院组装和存放的(http://www.academy.rpi.du/). 该服务器预计将在2003年春季或夏季转移到Voorhees计算中心。
未来发展方向
我们的目的是使RNAML输出保持最新并符合DTD文件:http://www-lbit.iro.umontreal.ca/rnaml/current/rnaml.dtd。在接下来的一两个月内,RNAML输出将包含“x个,年与“sir图”程序生成的结构图相对应的坐标(如果可用)。在未来的一两年里,我们将看到目前正在开发的重要、新颖的应用程序被纳入其中。
配分函数杂交
已经创建了新的算法来折叠和杂交两条分开的RNA或DNA链(41). 该软件的初始版本现在正在仔细地重新编程和更新。我们正在研究提高预测准确性的理论和实验方法。我们也开始考虑处理探针与芯片上固定的核酸杂交的热力学(42).
数据库搜索
我们正在开发一种新的“类BLAST”算法,用于搜索核酸数据库,该算法基于计算机科学散列技术,以提高速度,但搜索DNA或RNA两部分之间最佳互补的区域。评分基于RNA或DNA的最近邻自由能参数。现在,一个早期版本被用于在mRNA数据库中搜索micro-RNA(miRNAs)的假定结合位点,以及搜索基因特异性DNA探针的(不希望的)替代结合位点。
用户界面的开发
mfold web服务器的当前用户界面已经有好几年的历史了,无论是部分重写还是全部重写都会使其受益。通过表格提交数据仍然是合理的。可以很容易地添加用于上传序列文件的“浏览”选项。这将有助于更容易地输入约束信息。更有价值的是预处理器,它可以检查约束信息的一致性和合理性。到目前为止,由于约束中的小错误,许多受约束的折叠都失败了。
重新设计后,输出页面将大大受益。作为第一步,相对较新的“sir_graph”程序应取代旧的naview程序来创建二级结构图。最好创建交互式结构绘图功能。这种功能已经存在于“sir_graph”程序的独立版本中。我们有幸在RPI拥有尖端的电子媒体学院;一个可能与我们合作为web服务器创建真正一流的用户界面的小组。
命名MFOLD WEB服务器
使用mfold web服务器的作者应引用本文作为一般参考,还应包括入口页面的URL,http://www.bioinfo.rpi.edu/applications/mfold。web服务器页面将列出与所使用的自由能参数和底层软件相关的其他引文。
补充材料
补充材料可在NAR Online上获得,也可在以下网址找到MFOLDROOT/www-NAR03/supp公司.
致谢
这项工作在一定程度上得到了美国国立卫生研究院(National Institutes of Health)拨款#GM54250的支持。我感谢Gerald Johns在华盛顿大学服务器早期提供的硬件和系统支持。所有的图形应用程序(除了naview)都是由Darrin Stewart编写的,他还编写了用于交互式查看绘图的“cgi”脚本。我感谢Art Sanderson(RPI研究副总裁)将我与电子媒体学院联系起来,并支持这一项目;Bill Shumway,发起并促进与IBM Research的交互;以及Alex Yu,他在组装硬件、组织web服务器布局、移植应用程序以及保持服务器日复一日地运行方面做了大量工作。最后,我感谢IBM Research为我们提供的SUR拨款,它为我们提供了这一宝贵资源,使我们的计算机能力提高了三十倍。
图1。此图再现了RNA折叠的部分初始结果页面。在文本区域中复制了一些输入。各种超链接导致实际的折叠结果。
图2。通过将两个互补DNA序列与六个Ns连接,将其连接起来。这两个循环被标记为“1”和“2”。循环“1”是一个外部循环,这是正确的,但循环“2”是发夹循环而不是外部循环。
| G/A公司 | 转交 | A/C公司 | G/T公司 | G/C公司 | 自动转账 | 不是A | 不是C | 不是G | 不是T | 任何底座 |
| 对 | Y(Y) | M(M) | K(K) | S公司 | W公司 | B类 | D类 | H(H) | V(V) | N个 |
| G/A公司 | 转交 | A/C公司 | G/T公司 | G/C公司 | 自动转账 | 不是A | 不是C | 不是G | 不是T | 任何底座 |
| 对 | Y(Y) | M(M) | K(K) | S公司 | W公司 | B类 | D类 | H(H) | V(V) | N个 |
| G/A公司 | 转交 | A/C公司 | G/T公司 | G/C公司 | 自动转账 | 不是A | 不是C | 不是G | 不是T | 任何底座 |
| 对 | Y(Y) | M(M) | K(K) | S公司 | W公司 | B类 | D类 | H(H) | V(V) | N个 |
| G/A公司 | 转交 | A/C公司 | G/T公司 | G/C公司 | 自动转账 | 不是A | 不是C | 不是G | 不是T | 任何底座 |
| 对 | Y(Y) | M(M) | K(K) | S公司 | W公司 | B类 | D类 | H(H) | V(V) | N个 |
表2。jpg和png图像的分辨率和图像大小,单位分别为每平方英寸像素和像素
| 分辨率 | 60×60 | 72×72 | 110×110 | 200×200 | 250×250 | 300×300 |
| 图像大小 | 510×660 | 612×792 | 935×1210 | 1700×2200 | 2125×2750 | 2550×3300 |
| 分辨率 | 60×60 | 72×72 | 110×110 | 200×200 | 250×250 | 300×300 |
| 图像大小 | 510×660 | 612×792 | 935×1210 | 1700×2200 | 2125×2750 | 2550×3300 |
表2。jpg和png图像的分辨率和图像大小,单位分别为每平方英寸像素和像素
| 分辨率 | 60×60 | 72×72 | 110×110 | 200×200 | 250×250 | 300×300 |
| 图像大小 | 510×660 | 612×792 | 935×1210 | 1700×2200 | 2125×2750 | 2550×3300 |
| 分辨率 | 60×60 | 72×72 | 110×110 | 200×200 | 250×250 | 300×300 |
| 图像大小 | 510×660 | 612×792 | 935×1210 | 1700×2200 | 2125×2750 | 2550×3300 |
| 序列长度 | 编号增量 |
| 1–50 | 10 |
| 51–300 | 20 |
| >300 | 50 |
| 序列长度 | 编号增量 |
| 1–50 | 10 |
| 51–300 | 20 |
| >300 | 50 |
| 序列长度 | 编号增量 |
| 1–50 | 10 |
| 51–300 | 20 |
| >300 | 50 |
| 序列长度 | 编号增量 |
| 1–50 | 10 |
| 51–300 | 20 |
| >300 | 50 |
| δG公司 | 颜色 |
| ΔG公司=δG公司 | FFFFFF(黑色) |
| ΔG公司<δG公司≤ΔG公司+δδG公司/3 | D40721(红色) |
| ΔG公司+δδG公司/3<δG公司≤ΔG公司+2δG公司/3 | 12CC24(绿色) |
| ΔG公司+2δδG公司/3<δG公司≤ΔG公司+δδG公司 | B6C614(黄色) |
| δG公司 | 颜色 |
| ΔG公司=δG公司 | FFFFFF(黑色) |
| ΔG公司<δG公司≤ΔG公司+δδG公司/3 | D40721(红色) |
| ΔG公司+δδG公司/3<δG公司≤ΔG公司+2δδG公司/3 | 12CC24(绿色) |
| ΔG公司+2δδG公司/3<δG公司≤ΔG公司+δδG公司 | B6C614(黄色) |
| δG公司 | 颜色 |
| ΔG公司=δG公司 | FFFFFF(黑色) |
| ΔG公司<δG公司≤ΔG公司+δδG公司/3 | D40721(红色) |
| ΔG公司+δδG公司/3<δG公司≤ΔG公司+2δδG公司/3 | 12CC24(绿色) |
| ΔG公司+2δδG公司/3<δG公司≤ΔG公司+δδG公司 | B6C614(黄色) |
| δG公司 | 颜色 |
| ΔG公司=δG公司 | FFFFFF(黑色) |
| ΔG公司<δG公司≤ΔG公司+δδG公司/3 | D40721(红色) |
| ΔG公司+δδG公司/3<δG公司≤ΔG公司+2δδG公司/3 | 12CC24(绿色) |
| ΔG公司+2δδG公司/3<δG公司≤ΔG公司+δδG公司 | B6C614(黄色) |
表5。mfold web服务器是RPI-Wadsworth生物信息中心网站上的主要应用程序。它位于由双1组成的高性能web服务器上 带4个GHz Pentium处理器 Gb内存和73 Gb磁盘,连接到35个其他双1的群集 GHz奔腾,每一个有1个 Gb内存和36 Gb磁盘
| 描述 | 数量 |
| X系列330 1U单处理器 | 36 |
| #33l315 1个 Gb ecc Rdimm | 38 |
| 附加1 GHz奔腾处理器 | 36 |
| 平板显示器9511AG1 | 1 |
| 平板显示器机架套件 | 1 |
| 键盘28l3644 | 1 |
| 机架9306910 | 1 |
| 高清36 Gb驱动器37L7206 | 36 |
| DLT磁带机20/40 千兆字节 | 1 |
| Linux操作系统 | 1 |
| 100baseT以太网交换机 | 三 |
| APC Smart UPS 1400RMB(仅适用于web服务器) | 1 |
| 其他电缆和安装硬件 | 1 |
| 描述 | 数量 |
| X系列330 1U单处理器 | 36 |
| #第33页第315页 Gb ecc Rdimm | 38 |
| 附加1 GHz奔腾处理器 | 36 |
| 平板显示器9511AG1 | 1 |
| 平板显示器机架套件 | 1 |
| 键盘28l3644 | 1 |
| 机架9306910 | 1 |
| 高清36 Gb驱动器37L7206 | 36 |
| DLT磁带机20/40 千兆字节 | 1 |
| Linux操作系统 | 1 |
| 100baseT以太网交换机 | 三 |
| APC Smart UPS 1400RMB(仅适用于web服务器) | 1 |
| 其他电缆和安装硬件 | 1 |
表5。mfold web服务器是RPI-Wadsworth生物信息中心网站上的主要应用程序。它位于由双1组成的高性能web服务器上 带4个GHz Pentium处理器 Gb内存和73 Gb磁盘,连接到35个其他双1的群集 GHz奔腾,每一个有1个 Gb内存和36 Gb磁盘
| 描述 | 数量 |
| X系列330 1U单处理器 | 36 |
| #33l315 1个 Gb ecc Rdimm | 38 |
| 附加1 GHz奔腾处理器 | 36 |
| 平板显示器9511AG1 | 1 |
| 平板显示器机架套件 | 1 |
| 键盘28l3644 | 1 |
| 机架9306910 | 1 |
| 高清36 Gb驱动器37L7206 | 36 |
| DLT磁带机20/40 千兆字节 | 1 |
| Linux操作系统 | 1 |
| 100baseT以太网交换机 | 三 |
| APC Smart UPS 1400RMB(仅适用于web服务器) | 1 |
| 其他电缆和安装硬件 | 1 |
| 描述 | 数量 |
| X系列330 1U单处理器 | 36 |
| #第33页第315页 Gb ecc Rdimm | 38 |
| 附加1 GHz奔腾处理器 | 36 |
| 平板显示器9511AG1 | 1 |
| 平板显示器机架套件 | 1 |
| 键盘28l3644 | 1 |
| 机架9306910 | 1 |
| 高清36 Gb驱动器37L7206 | 36 |
| DLT磁带机20/40 千兆字节 | 1 |
| Linux操作系统 | 1 |
| 100baseT以太网交换机 | 三 |
| APC Smart UPS 1400RMB(仅适用于web服务器) | 1 |
| 其他电缆和安装硬件 | 1 |
工具书类
1Doty,P.、Boedtker,H.、Fresco,J.R.、Haselkorn,R.和Litt,M(
1959
)核糖核酸的二级结构。程序。美国国家科学院。科学。美国
,45
,482
–499. 2Fresco,J.R.、Alberts,B.M.和Doty,P(
1960
)核糖核酸二级结构的一些分子细节。自然
,188
,98
–101. 三。Borer,P.N.,Dengler,B.,Tinoco,I.,Jr和Uhlenbeck,O.C(
1974
)核糖核酸双链螺旋的稳定性。分子生物学杂志。
,86
,843
–853. 4Tinoco,I.,Jr和Uhlenbeck,O.C(
1971
)核糖核酸二级结构的估计。自然
,230
,362
–367. 5Tinoco,I.,Jr,Borer,P.N.,Dengler,B.,Levine,M.D.,Uhlenbeck,O.C.,Crothers,D.M.和Gralla,J(
1973
)改进了对核糖核酸二级结构的估计。自然新生物。
,246
,40
–41. 6O.C.Uhlenbeck,P.N.Borer,Dengler,B.和Tinoco,I.Jr(
1973
)RNA发夹环的稳定性:A6-C类米-单位6.分子生物学杂志。
,73
,483
–496. 7Delisi,C.和Crothers,D.M(
1971
)RNA二级结构预测。程序。美国国家科学院。科学。美国
,68
,2682
–2685. 8Waterman,M.S.和Smith,T.M(
1978
)RNA二级结构:完整的数学分析。数学。Biosci公司。
,42
,257
–266. 9沃特曼医学博士(
1978
)单链核酸的二级结构。在罗塔,G.-C.(编辑)中,基础和组合数学研究在数学进展、补充研究中排名第一。
纽约学术出版社,pp。167
–212. 10Nussinov,R.、Pieczenik,G.、Griggs,J.R.和Kleitman,D.J(
1978
)循环匹配算法。SIAM J.应用。数学。
,35
,68
–82. 11Nussinov,R.和Jacobson,A.B(
1980
)预测单链RNA二级结构的快速算法。程序。美国国家科学院。科学。美国
,77
,6309
–6313. 12Zuker,M.和Stiegler,P(
1981
)利用热力学和辅助信息优化大RNA序列的计算机折叠。核酸研究。
,9
,133
–148. 13Sankoff,D.,Kruskal,J.B.,Mainville,S.和Cedergren,R.J(
1983
)确定含有多个环的RNA二级结构的快速算法。第三章。Sankoff,D.和Kruskal,J.B.(编辑),时间扭曲、字符串编辑和大分子:序列比较的理论与实践
艾迪森·韦斯利·雷丁,马萨诸塞州,pp。93
–120. 14M.祖克(
1989
)RNA结构的计算机预测。方法酶制剂。
,180
,262
–288. 15Devereux,J.、Haeberli,P.和Smithies,O(
1984
)VAX的一整套序列分析程序。核酸研究。
,12
,387
–395. 16西萨瑟(
1977
)球蛋白mRNA序列:碱基配对和进化意义分析。冷泉港交响乐团。数量。生物。
,42
,985
–1002. 17Freier,S.M.、Kierzek,R.、Jaeger,J.A.、Sugimoto,N.、Caruthers,M.M.、Neilson,T.和Turner,D.H(
1986
)用于预测RNA双链稳定性的改进自由能参数。程序。美国国家科学院。科学。美国
,83
,9373
–9377. 18M.祖克(
1989
)找到RNA分子的所有次优折叠。科学类
,244
,48
–52。 19Jaeger,J.A.、Turner,D.H.和Zuker,M(
1989
)改进了对RNA二级结构的预测。程序。美国国家科学院。科学。美国
,86
,7706
–7710. 20Jaeger,J.A.、Turner,D.H.和Zuker,M(
1990
)预测RNA的最佳和次最佳二级结构。方法酶制剂。
,183
,281
–306. 21M.祖克(
1994
)通过能量最小化预测RNA二级结构。第23章。在A.M.格里芬和H.G.格里芬(编辑),序列数据的计算机分析
,25
第二部分,Humana出版社,新泽西州Totowa,pp。267
–294. 22M.Zuker、D.H.Mathews和D.H.Turner(
1999
)RNA二级结构预测的算法和热力学:实用指南。在Barciszewski,J.和Clark,B.F.C.(编辑)中,RNA生物化学和生物技术,北约科学伙伴关系子系列第70名:3:高科技
第二章,荷兰多德雷赫特Kluwer学术出版社,pp。11
–43. 23D.H.马修斯、T.C.安德烈、J.金、D.H.特纳和M.祖克(
1998
)用改进的自由能参数预测RNA二级结构的更新递归算法。第15章。在Leontis,N.B.和SantaLucia,J.,Jr(编辑),美国化学学会研讨会系列682
,美国化学学会华盛顿特区,pp。246
–257. 24D.H.马修斯、D.H.特纳和M.祖克(
2000
)RNA二级结构预测。第11.2章。在Beaucage,S.、Bergstrom,D.E.、Glick,G.D.和Jones,R.A.(编辑),当前核酸化学协议
约翰·威利父子公司,纽约州纽约市,pp。1
–10. 25Walter,A.E.,Turner,D.H.,Kim,J.,Lyttle,M.H.,Muöller,P.,Mathews,D.H..和Zuker,M(
1994
)螺旋的同轴堆积增强了寡核苷酸的结合并改善了RNA折叠的预测。程序。美国国家科学院。科学。美国
,91
,9218
–9222. 26D.H.马修斯、J.萨宾娜、M.祖克和D.H.特纳(
1999
)热力学参数的扩展序列依赖性提高了对RNA二级结构的预测。分子生物学杂志。
,288
,911
–940。 27小圣卢西亚(
1998
)聚合物、哑铃和寡核苷酸DNA近邻热力学的统一观点。程序。美国国家科学院。科学。美国
,95
,1460
–1465. 28H.T.阿拉维和J.圣卢西亚(
1997
)DNA内部G·T失配的热力学和NMR。生物化学
,36
,10581
–10594. 29H.T.阿拉维和J.圣卢西亚(
1998
)DNA内部G·A失配的最近邻热力学参数。生物化学
,37
,2170
–2179. 30H.T.阿拉维和J.圣卢西亚(
1998
)DNA内部C·T失配的热力学。核酸研究。
,26
,2694
–2701. 31H.T.阿拉维和J.圣卢西亚(
1998
)DNA内部A·C失配的最近邻热力学:序列依赖性和pH效应。生物化学
,37
,9435
–9444。 32Peyret,N.、Senevirtne,P.A.、Allawi,H.T.和SantaLucia,J.小(
1999
)具有内部A·A、C·C、G·G和T·T失配的DNA序列的最近邻热力学和NMR。生物化学
,38
,3468
–3477. 33Bommarito,S.、Peyret,N.和SantaLucia,J.,Jr(
2000
)末端悬垂的DNA序列的热力学参数。核酸研究。
,28
,1929
–1934. 34北卡罗来纳州佩雷特(
2000
)核酸杂交预测:参数和算法。塞利斯博士,韦恩州立大学化学系,密歇根州底特律。 35Cornish-Bowden,A(
1985
)核酸序列中不完全指定碱基的命名:1984年建议。核酸研究。
,13
,3021
–3030. 36Zuker,M.和Jacobson,A(
1998
)使用可靠性信息注释RNA二级结构。核糖核酸
,4
,669
–679. 37Waugh,A.、Gendron,P.、Altman,R.、Brown,J.W.、Case,D.、Gautheret,D.、Harvey,S.C.、Leontis,N.、Westbrook,J.、Westhof,E。等。(
2002
)RNAML:交换RNA信息的标准语法。核糖核酸
,8
,707
–717. 38Zuker,M.和Jacobson,A.G(
1995
)RNA二级结构预测中的“确定良好”区域。应用于大小亚单位rRNA。核酸研究。
,23
,2791
–2798. 39Bruccoleri,R.和Heinrich,G(
1988
)一种改进的核酸二级结构显示算法。计算。申请。Biosci公司。
,4
,167
–173. 40Rouillard,J.-M.,Herbert,C.J.和Zuker,M(
2002
)寡核苷酸阵列:用于微阵列的基因组级寡核苷酸设计。生物信息学
,18
,486
–487. 41R.A.Dimitrov和M.Zuker(
2003
)预测双链核酸的杂交和熔化。生物物理学。J。
,正在印刷中。 42Fotin,A.V.、Drobyshev,A.L.、Proudnikov,D.Y.、Perov,A.N.和Mirzabekov,A.D(
1998
)寡脱氧核苷酸微芯片双工的并行热力学分析。核酸研究。
,26
,1515
–1521.








{kind=link}
{kind=link}