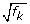假设情景
估计的标准化因子应确保在两个样本中具有相同表达水平的基因不会被检测为DE。为了进一步强调RNA-seq数据中需要更复杂的标准化程序,请考虑一个简单的思维实验。假设我们有一个比较两个RNA群体a和B的测序实验。在这个假设场景中,假设在B中表达的每个基因在a中表达的转录物数量相同。然而,假设样品A还包含一组数量和表达相同的基因,而这些基因在B中没有表达。因此,样品A的总表达基因是样品B的两倍,也就是说,其RNA产量是样品B大小的两倍。如果没有任何额外的调整,两个样本中表达的基因的平均读取次数将是样本a的一半,因为读取的基因数是样本a基因数的两倍。因此,正确的归一化将样本A调整为2倍。
上述假设示例强调了这样一个概念,即库中特定基因的读取比例取决于整个样本的表达特性,而不仅仅是该基因的表达水平。显然,上面的例子是人为的。然而,在生物甚至技术情况下,需要进行这种规范化。例如,如果RNA样本被污染,表示污染的读数将从真实样本中删除读数,从而减少感兴趣的读数数量,并抵消每个基因的比例。然而,正如我们所证明的,样本之间RNA组成的真正生物差异将是归一化的主要原因。
采样框架
规范化需求的更正式解释使用以下框架Y(Y)
gk公司
作为基因的观察计数克在库中k个从原始阅读中总结,μ
gk公司
作为真实和未知的表达水平(转录本数量),L(左)
克
作为基因的长度克和N个
k个
作为库的读取总数k个。我们可以对Y(Y)
gk公司
作为:
S公司
k个
表示样本的总RNA输出。RNA-seq数据分析背后的问题是N个
k个
已知的是,S公司
k个
未知,根据RNA组成,不同样品之间可能会有很大差异。如上所述,如果一个群体的总RNA输出量较大,那么相对于另一个样本,RNA-seq实验将对许多基因进行欠采样。
在这个阶段,我们将上述模型中的方差保留为Y(Y)
gk公司
未指定。根据实验情况,泊松似乎适用于技术复制[6,7]负二项式可能适用于从生物复制中观察到的额外变异[14]. 同样值得注意的是,在实践中L(左)
克
通常被吸收到μ
gk公司
参数,并且不在推理过程中使用。然而,已经很好地证明,基因长度偏差在基因表达分析中非常突出[15].
M值归一化方法的修剪平均值
总RNA产量,S公司
k个
不能直接估计,因为我们不知道每个基因的表达水平和真实长度。然而,两个样本的相对RNA产量,(f)
k个
=S
k个
/秒
k’
基本上是一个全球褶皱变化,可以更容易地确定。我们提出了一种经验策略,在假设大多数样本不是DE的情况下,将样本之间的基因总表达水平等同。一种简单而稳健的估算RNA生成比率的方法是使用对数表达比率的加权修剪平均数(修剪平均数M值(TMM))。对于测序数据,我们将基因的对数变换定义为:
和绝对表达水平:
为了稳妥地总结观察到的M值,我们在取加权平均值之前对M值和A值进行了修剪。精度(方差的倒数)权重用于解释这样一个事实,即具有较大读取计数的基因的对数倍变化(实际上是对数相对风险)在对数尺度上的方差较低。有关详细信息,请参见材料和方法。
对于两个样本的比较,只有一个相对比例因子((f)
k个
)是必需的。它可用于调整两个库大小(将参考除以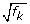 并将非引用乘以)在统计分析中(例如,Fisher精确检验;有关更多详细信息,请参阅材料和方法)。
并将非引用乘以)在统计分析中(例如,Fisher精确检验;有关更多详细信息,请参阅材料和方法)。
通过选择一个样本作为参考并计算每个非参考样本的TMM因子,可以计算多个样本的归一化因子。与双样本比较类似,TMM归一化因子可以构建到用于测试DE的统计模型中。例如,泊松模型会将观察到的库大小修改为有效的库大小,从而调整建模的平均值(例如,在广义线性模型中使用额外的偏移量;有关详细信息,请参见材料和方法)。
肝脏与肾脏数据集
我们将我们的方法应用于公开可用的转录谱数据集,比较肝脏和肾脏RNA来源的几个技术复制[6]. 图1a个显示了在计算读取总数的标准归一化程序后,肾脏样品的两个技术复制品之间的M值分布。这些技术复制品的M值分布集中在零附近。然而,图1亿显示肝脏和肾脏样本之间的对数比显著地向肾脏中更高的表达偏移,即使在考虑读取总数之后也是如此。另一个突出显示的(绿线)是一组家政基因的观察到的M值的分布,显示出显著偏离零。如果按比例缩放到适当标准化RNA-seq数据的读取总数,则预期不会出现这种对数变换偏移。这种偏见的解释很简单。图中的M与A图1c个说明肝脏中存在一组显著的高表达基因(黑箭头)。因此,M值的分布(肝脏到肾脏)呈负向倾斜。由于大量测序都是针对这些肝脏特异性基因的,因此剩余基因的测序较少,因此M值(因此DE值)成比例地扭曲为肾特异性。
对这对样品应用TMM归一化后,归一化因子为0.68(对数2标度为-0.56;如图中的红线所示1b、c),反映了大多数肝脏基因的采样不足。TMM因子对于覆盖率较低的数据是稳健的,在这些数据中,可能会有更多的零计数基因(附加文件中的图S1a1)并且对于配平参数的合理值是稳定的(附加文件中的图S1b1). 在DE的统计测试中使用TMM归一化(见材料和方法)会导致肝脏(47%)和肾脏(53%)中相似数量的基因显著增加。相比之下,标准规范化(到最初用于[6])导致大多数DE基因在肾脏中显著较高(77%)。值得注意的是,在TMM归一化后,使用标准归一化方法鉴定为DE的基因中仍检测到不到70%(表1). 此外,我们还发现了大量家政基因的对数变换(来自[16])平均而言,从零开始的偏移非常接近估计的TMM因子,从而使我们的稳健估计程序具有可信度。此外,使用未调整的测试程序,肝脏和肾脏中分别有8%和70%的管家基因显著上调。TMM调整后,DE管家基因的比例分别变为26%和41%,这是一个较低的总数,并且两个组织之间更加对称。当然,假设微阵列数据已经适当归一化(附加文件中的图S21). 综上所述,这些结果表明RNA-seq数据规范化的关键作用。
表1使用不同的归一化方法,以<0.001的错误发现率在肝脏和肾脏之间差异表达的基因数量 其他数据集
在其他RNA-seq数据集中,由RNA组成差异引起的对数变换的全球变化在不同程度上发生。例如,克隆人的M与A情节等. [12]数据集(附加文件中的图S31)给出了在SOLiD™系统上测序的两个样本(类胚体与胚胎干细胞)之间的估计TMM比例因子1.04。该数据集的M与A图也突出了一组有趣的基因,这些基因总体表达较低,但在类胚体中表达较高。这解释了剩余基因对数变换的正向变化。TMM比例因子似乎接近一组大约500个小鼠家政基因(来自[17]). 另一个例子是,李等. [18]使用llumina 1G基因组分析仪的数据集显示出对数变化的总体分布发生了变化,并给出了0.904的TMM比例因子(附加文件中的图S41). 然而,有些基于序列的数据集具有非常相似的RNA输出,可能不需要进行重大调整。例如,来自Kuchenbauer的小型RNA-seq数据等. [19]在log-fold-changes中只显示了适度的偏差(附加文件中的图S51).
尖峰控制有可能用于标准化。在这种情况下,将少量已知量的外来生物体RNA以特定浓度添加到每个样本中。为了使用加标对照进行归一化,在整个实验过程中,加标与样品的浓度比必须保持恒定。在实践中,这很难实现,并且小的变化将导致归一化因子的有偏估计。例如,使用来自Mortazavi的插入DNA等.数据集[11]将导致不切实际的标准化因子估计(附加文件中的图S61). 与微阵列一样使用实验数据仔细估计归一化因子通常更稳健(例如[20]).
模拟研究
为了研究TMM归一化方法的适用范围,我们开发了一个模拟框架来研究RNA组成对RNA-seq数据DE分析的影响。首先,我们模拟来自两个库的数据。我们包括每个样本唯一表达的基因数量参数,以及样本间差异表达基因的比例、大小和方向参数(见材料和方法)。图2a个显示了包含独特基因和DE基因的典型模拟的M与A图。通过模拟不同的总RNA输出,大多数非DE基因具有从零开始偏移的对数变换。在这种情况下,使用TMM归一化来解释潜在的RNA组成,可以减少使用Fisher精确测试的错误检测次数(图2亿). 在广泛的仿真参数范围内多次重复仿真,当比较仿真的真实归一化因子与使用TMM归一化估计的那些因子时,我们发现非常一致(附加文件中的图S71).
为了进一步比较TMM归一化与之前在RNA-seq数据DE分析中使用的方法的性能,我们扩展了上述模拟以包括重复测序运行。具体来说,我们比较了三种已发布的方法:经过对数转换的长度规范化计数数据和由Cloonan实现的分位数规范化等. [12],泊松回归[6]具有库大小、TMM归一化和泊松精确测试[8]具有库大小和TMM规范化。我们没有直接与Balwierz提出的标准化进行比较等. [13]因为肝脏和肾脏数据集似乎不遵循幂律分布,并且具有非常明显的计数分布(附加文件中的图S81). 此外,鉴于我们观察到的RNA组成偏差,尚不清楚等值样本的计数分布是否是最合理的程序。此外,我们没有直接将归一化与虚拟长度进行比较[2]或RPKM[11]归一化,因为没有提到对转换数据的统计分析。然而,我们用M与A图说明,它们的归一化并不能完全消除RNA组成偏差(附加文件中的图S9和S101).
在模拟中,我们使用了基因长度和计数的经验联合分布,因为克隆人等.过程同时需要这两个参数。我们将模拟数据泊松分布以模拟技术复制(附加文件中的图S111). 图3a年显示了两种情况下常见的基因之间的错误发现图,其中我们在第一种情况下引入了10%的唯一组表达,在两倍水平上引入了5%的DE,其中80%在第一种条件下更高。使用为微阵列数据开发的方法学的方法学表现一致较差,正如人们可能预期的那样,因为这些方法的分布假设完全不同。在其余方法(泊松似然比统计、泊松精确统计)中,性能非常相似;同样,TMM规范化对这两者都有显著的改进。