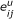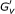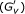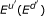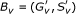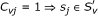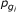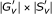给定表达式矩阵E类带有基因G公司={克1,克2,克三,...,克
n个
}和样品S公司={秒1,秒2,秒三,...,秒
米
}双星簇定义为b条= (G’,S’)其中G’ ⊂ G公司是基因的子集S’ ⊂ S公司是样本的子集。DeBi识别功能相干双团簇B={b条1,b条2,b条三,...,b条
我
}分为三个步骤。下面我们详细描述每个步骤。DeBi算法概述如图所示6DeBi算法基于一种众所周知的数据挖掘方法,称为最大频繁项集[28]. 我们将其称为最大频繁基因集,如我们的问题定义所示。算法的伪代码在附加文件中1.
前期工作
输入的基因表达数据根据上调或下调进行二值化。让E类u个和E类d日分别表示上下调节二进制矩阵。然后是条目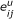 属于E类u个定义如下:
属于E类u个定义如下:
和条目 属于E类d日与c折下调截止线类似定义。折变截止线c(c)通常设置为2。
属于E类d日与c折下调截止线类似定义。折变截止线c(c)通常设置为2。
用最大频繁基因集算法寻找种子双聚类
DeBi算法通过迭代应用最大频繁基因集算法来识别种子基因集。我们首先定义术语支持,我们稍后将在算法中使用。这个支持基因的克
我
,我=1,...,n个,定义如下:
换句话说支持是基因向量的样本比例e(电子)
我
.为1。这进一步扩展到基因集。让 成为v(v)第基因集。对于一组基因向量,我们定义了它们的表型载体C
v(v)
作为它们的元素逻辑AND:
成为v(v)第基因集。对于一组基因向量,我们定义了它们的表型载体C
v(v)
作为它们的元素逻辑AND:
这个支持然后将基因集的部分定义为表型载体为1的样品的部分。
基因集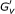 是(c(c)
1
,c(c)
2
) -频繁的iff支持支持
是(c(c)
1
,c(c)
2
) -频繁的iff支持支持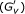 大于c(c)
1
和基数
大于c(c)
1
和基数 在上面c(c)
2
.何时c(c)
1
和c(c)
2
我们将简单地说一个频繁的基因集。基因集是最频繁的如果它是频繁的,并且它没有超集是频繁的。
在上面c(c)
2
.何时c(c)
1
和c(c)
2
我们将简单地说一个频繁的基因集。基因集是最频繁的如果它是频繁的,并且它没有超集是频繁的。
检测最大频率基因集的最简单方法是蛮力方法,其中每个可能的子集G公司={克1,克2,克三,...,克
n个
}是一个候选频繁集。为了找到频繁集,我们计算每个候选集的支持度。MAFIA算法是一种高效的实现,用于查找支持高于给定阈值的最大频繁集[28]. MAFIA的搜索策略利用有效的剪枝技术对基因集格进行深度优先遍历。它通过单调性原则避免了对所有候选基因集的穷举。单调性原理表明频繁项集的每个子集都是频繁的。它使用此属性修剪具有罕见子模式的候选。
在DeBi算法的第一步中,将MAFIA迭代应用于二进制矩阵,依次降低支持阈值。最初,MAFIA应用于完整的二进制矩阵E类u个(E类d日)具有支持值(c(c)
1
)
0
等于支持度最高的基因的支持值。迭代中k个,MAFIA的支持值阈值为 将识别出的最大频繁集添加到种子基因集中B以及B从二进制矩阵中删除E类u个(E类d日). 在每次迭代中,MAFIA应用于修改后的矩阵
将识别出的最大频繁集添加到种子基因集中B以及B从二进制矩阵中删除E类u个(E类d日). 在每次迭代中,MAFIA应用于修改后的矩阵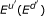 重复该过程,直到用户定义最小支撑已达到参数。
重复该过程,直到用户定义最小支撑已达到参数。
扩展和过滤双簇
在DeBi的第二步中,确定的种子基因集 使用本地搜索进行扩展。对于每个双集群
使用本地搜索进行扩展。对于每个双集群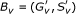 ,v(v)=1,。。。,我,我们有二元表型载体C类
v(v)
=∧(e(电子)1,...,e(电子)
k个
) = (C类v(v)1,...,C类
虚拟机
). 的条目C类
v(v)
指示双团簇样品的指数。如果
,v(v)=1,。。。,我,我们有二元表型载体C类
v(v)
=∧(e(电子)1,...,e(电子)
k个
) = (C类v(v)1,...,C类
虚拟机
). 的条目C类
v(v)
指示双团簇样品的指数。如果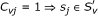 ,j个= 1,...,米即样品秒
j个
属于双星团b
v(v)
.基因克
我
,我=1,...,n个,是基因集的一个元素如果e(电子)
我
.与关联C类
v(v)
我们在2×2列联表上使用Fisher精确检验评估了一个双簇的表型载体与另一个基因之间的关联强度。列联表中的单元格计算包含1或0的表型向量和包含1或1的基因向量的四种可能性发生的频率。然后,Fisher精确检验检验列联表中以及两个向量之间的独立性。
,j个= 1,...,米即样品秒
j个
属于双星团b
v(v)
.基因克
我
,我=1,...,n个,是基因集的一个元素如果e(电子)
我
.与关联C类
v(v)
我们在2×2列联表上使用Fisher精确检验评估了一个双簇的表型载体与另一个基因之间的关联强度。列联表中的单元格计算包含1或0的表型向量和包含1或1的基因向量的四种可能性发生的频率。然后,Fisher精确检验检验列联表中以及两个向量之间的独立性。
A基因克
我
,我=1,...,n个添加到基因集如果pvalue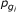 Fisher精确测试返回的值低于参数α。它从中删除b条
v(v)
如果概率高于α并添加到b条
v(v)
如果概率小于α对于此过程,关联概率需要计算每个基因的双聚类。然而,我们利用超几何分布的单调性减少了计算量。我们预先计算列联表条目的截止值,这些条目产生的p值略高于α.让σ
1,英寸
和σ
1,输出
分别表示基因向量在双簇样本中的1个数和基因向量在两簇样本外的1个数。我们发现最小值σ
1,英寸
和最大σ
1,输出
在这个边界。然后,我们只对那些具有σ
1,英寸
>最小σ
1,英寸
和σ1,OUT(输出)<最大σ1,OUT(输出).
Fisher精确测试返回的值低于参数α。它从中删除b条
v(v)
如果概率高于α并添加到b条
v(v)
如果概率小于α对于此过程,关联概率需要计算每个基因的双聚类。然而,我们利用超几何分布的单调性减少了计算量。我们预先计算列联表条目的截止值,这些条目产生的p值略高于α.让σ
1,英寸
和σ
1,输出
分别表示基因向量在双簇样本中的1个数和基因向量在两簇样本外的1个数。我们发现最小值σ
1,英寸
和最大σ
1,输出
在这个边界。然后,我们只对那些具有σ
1,英寸
>最小σ
1,英寸
和σ1,OUT(输出)<最大σ1,OUT(输出).
在最后一步中,我们转向双星团之间有时非常复杂的重叠结构。目标是过滤这组双簇,使剩余的双簇很大,只重叠很少。双聚类的大小被定义为基因的数量乘以双聚类中样本的数量,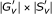 当两个双聚类共享共同的样本和基因时,它们会重叠。重叠的大小是常见样本数和常见基因数的乘积。为了筛选出大部分包含在较大的双簇中的双簇,我们从最大的双簇开始,并将其与其他双簇进行比较。那些与最大的重叠超过较小的大小的L%(通常为50%)的双簇将被删除。然后从剩下的第二大双星簇开始重复此操作,依此类推。
当两个双聚类共享共同的样本和基因时,它们会重叠。重叠的大小是常见样本数和常见基因数的乘积。为了筛选出大部分包含在较大的双簇中的双簇,我们从最大的双簇开始,并将其与其他双簇进行比较。那些与最大的重叠超过较小的大小的L%(通常为50%)的双簇将被删除。然后从剩下的第二大双星簇开始重复此操作,依此类推。
选择最佳alpha参数
为以下各项制定最佳标准α一个需要对一组双聚类的质量进行内在度量。为此,对于双星团v(v),我们定义其分数我
v(v)
作为包含基因的对数p值的负和,其中个体对
克
是Fisher精确测试的p值:
然而,这个双聚类得分我
v(v)
取决于双聚类的大小(基因数×条件数),为了使其在双聚类之间具有可比性,需要校正其大小。我们通过随机程序计算预期的双聚类得分。生成了大量随机表型向量,例如500个,与双簇具有相同数量的1s。对于这些随机表型载体,计算关于双聚类中每个基因的Fisher精确测试p值。一个人随机获得我
v(v)
通过在双簇基因上添加对数p值来得分。这些随机双聚类得分的平均值是期望的估计值。最后,归一化镍
v(v)
分数通过除法确定我
v(v)
通过这个估计平均值和总的双聚类得分加拿大定义为镍
v(v)
所有发现的双聚类的归一化得分 。此分数用于区分不同的选择α。程序运行于α={10-2, 10-3,..., 10-100}我们选择α使CS最大化。
。此分数用于区分不同的选择α。程序运行于α={10-2, 10-3,..., 10-100}我们选择α使CS最大化。