在药物发现中,基于所谓经典力场的方法已被常规使用,并被认为是有用的。然而,人们也普遍认识到,力场模型中缺少一些重要的物理,导致适用性有限。
例如,在比较具有不同形式电荷的两个分子时,力场模型不能提供准确的预测,因为模型中没有考虑蛋白质极化。此外,由于缺乏形成和破坏化学键的能力,这些模型不适用于共价药物分子。
许多人都同意,基于量子力学的模拟是解决这些问题的方法,而量子力学是控制微观世界的基本物理。但量子力学模拟被认为成本太高,耗时太长,不实用。
Q模拟最近宣布推出蜂巢-G这使得基于量子力学的自由能微扰(FEP)模拟具有前所未有的吞吐量。对于每GPU卡每天100+纳秒的吞吐量,这些模拟将在几个小时内完成。直接应用基于量子力学的模拟来识别新药分子将改变计算机辅助药物发现。
这篇文章解释了QSimulate是如何通过开发创新算法并将其应用到软件中来应对这一挑战的,该软件利用了NVIDIA提供的最新GPU硬件,例如NVIDIA A100公司和NVIDIA H100公司。经过严格优化的软件利用了最近在CUDA工具包,包括中的条件图节点CUDA图(引入于版本12.3并在中增强版本12.4).
量子力学的吞吐量挑战
药物发现模拟的一个重要方面是,每次能量和力量评估都必须在几毫秒左右执行。这一要求源于这样一个事实,即由于蛋白质是柔性的,并且随着时间的推移会改变其形状,因此有必要通过分子动力学(MD)模拟在多纳秒内对热力学系综进行采样,以准确预测其性质。一个例子是药物分子与蛋白质靶点的结合亲和力。
如果目标是实现每天100纳秒的动态吞吐量(标准时间步长为2飞秒),则每个时间步长必须在不到2毫秒的壁时间内完成。经典的力场模拟已被证明可以实现这一吞吐量。例如,请参阅有关GROMACS公司和NAMD公司.
传统上,人们认为,即使使用现代NVIDIA GPU硬件,也很难(如果不是不可能)将量子力学模拟加速到毫秒/时间步长的状态。这就是为什么大多数现有的GPU上的量子力学模拟实现都专注于加速大型计算(例如耦合簇计算和大规模密度泛函理论计算)通常是几小时到几分钟或几天到几小时。要了解更多信息,请参阅GPU加速量子化学与分子动力学.
虽然有用,但这些方法并不直接适用于药物发现的自由能计算,因为吞吐量降低了几个数量级。从技术上讲,量子力学的吞吐量挑战源于这样一个事实,即量子力学本质上是一个具有大量参数的优化问题,必须迭代求解。
这不可避免地引入了复杂的控制逻辑并限制了并发程度。如果天真地实现,量子力学的复杂逻辑会导致频繁的设备到主机通信,从而降低GPU的利用率。
QUELO-G利用CUDA图和紧束缚量子力学方法克服了这一挑战(GFN-xTB)和混合量子力学/分子力学(QM/MM)方案.
使用CUDA图和条件节点的技术解决方案
考虑Krylov子空间算法作为迭代量子力学算法的一个例子。该方法构造了一个由试探向量组成的子空间,在该子空间中,从上一次迭代的最佳解中生成一个新的试探向量。迭代该过程,直到满足收敛标准。图1显示了算法的示意图。
 图1。A类 在GPU上使用CPU上的控制代码实现迭代求解器的标准方法(左),与使用条件图节点功能基于CUDA图的方法(右)相比
图1。A类 在GPU上使用CPU上的控制代码实现迭代求解器的标准方法(左),与使用条件图节点功能基于CUDA图的方法(右)相比
实现此算法的标准方法是让主机负责循环和条件分支,同时将所有其他部分卸载到设备。试验向量(以及子空间)可以保存在设备存储器中。然而,对于在主机上执行的控制逻辑,在每次迭代中,需要将残余误差(标量值)从设备传递到主机。
即使对于标量变量,设备到主机通信的延迟也远远不能忽略。由于底层算法的结构,这种情况会反复发生,这对于基于量子力学的动力学模拟的整体性能来说非常重要,同时还可以实现毫秒/时间步长的吞吐量。
QUELO-G中的软件实现使用了最近在CUDA Graphs中引入的条件图节点(自CUDA 12.3以来)。使用此功能,量子力学算法中的整个迭代过程可以映射到CUDA图,其中循环和条件分支在设备中执行(图2)。这样就无需在每次迭代中将标量从设备传输到主机。这种方法不仅可以显著提高性能,而且由于更好的抽象,还可以简化代码结构。
 图2。CUDA图表示量子力学波函数优化的QUELO-G算法。每一列都对应于一个条件节点的主体
图2。CUDA图表示量子力学波函数优化的QUELO-G算法。每一列都对应于一个条件节点的主体
此外,通过使用CUDA Graphs抽象代码,软件将自动从CUDA Graph运行时的进一步开发和优化中受益,该运行时适用于现有NVIDIA A100和NVIDIA H100 GPU以及未来的NVIDIA-GPU。
使用上面的实现策略,QM/MM每天的动态吞吐量超过100纳秒。由于QSimulate的生产平台旨在将每次模拟的成本降至最低,多仪表GPU(MIG)通常用于在GPU上执行七个并发动力学模拟。图3汇编了在A100和H100 GPU上测量的单位细胞中由约25K经典原子组成的蛋白-甘露系统的吞吐量。
已经有了A100,用于模拟由74个原子组成的量子力学区域的QM/MM MD吞吐量每天超过100纳秒,这是小分子药物的典型尺寸。对于一个H100,QM/MM MD和FEP吞吐量分别测量为120和90纳秒。在由200个原子组成的更大量子力学区域中,观察到A100和H100之间的加速比超过50%。请注意,这些定时可以通过未来的创新和软件优化来进一步改进。
 图3。在NVIDIA A100和H100 GPU上测量的QM/MM MD(绿色)和FEP(蓝色)模拟吞吐量。MIG用于执行七个并发动力学仿真
图3。在NVIDIA A100和H100 GPU上测量的QM/MM MD(绿色)和FEP(蓝色)模拟吞吐量。MIG用于执行七个并发动力学仿真
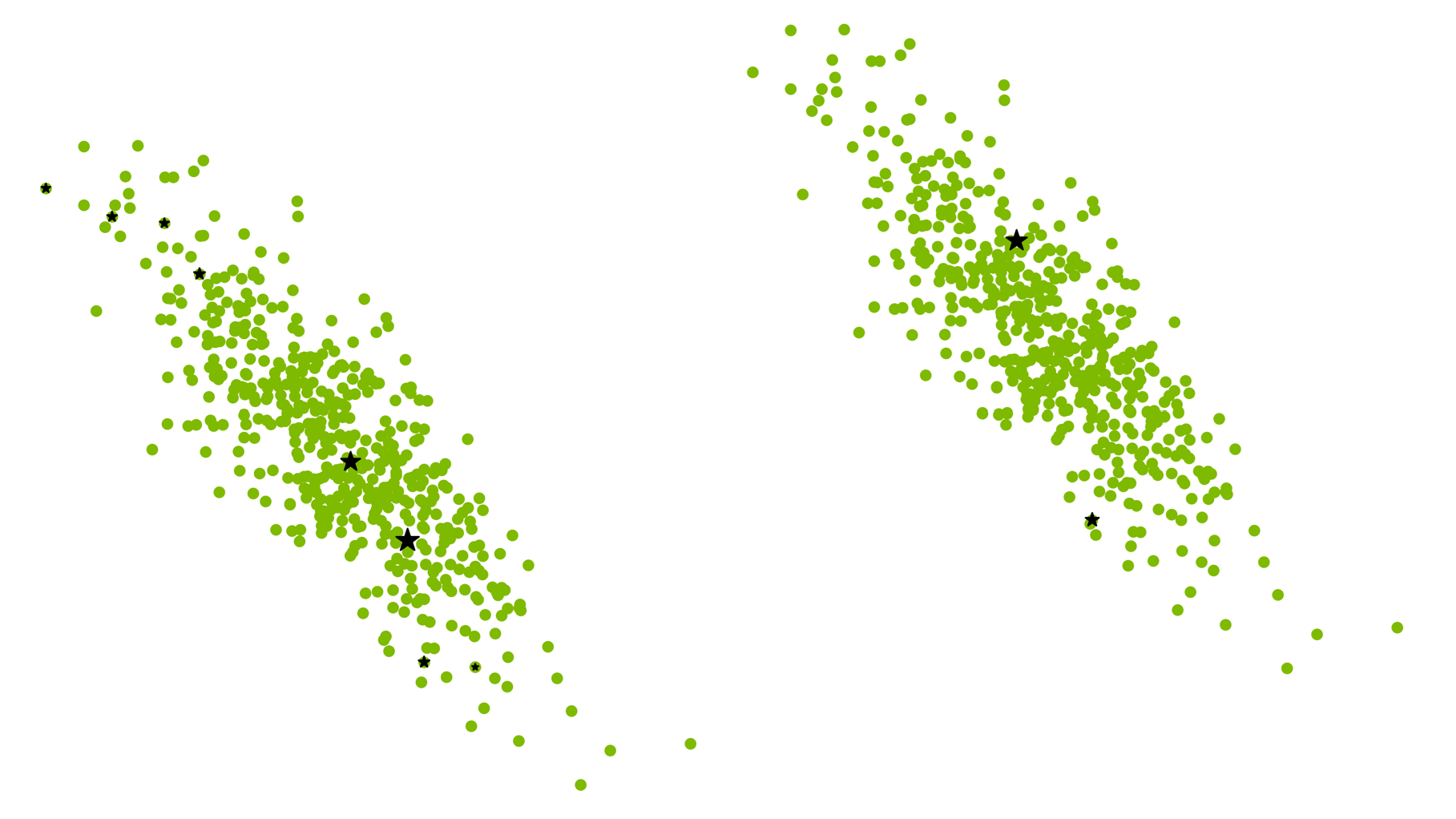
这些结果与QSimulate团队与RIKEN的Sugita集团图4显示了结果。图4中的四列对应于图3中的四个量子力学区域。CPU和GPU实现中的底层算法是相同的,包括截止值、阈值和参数。由于CPU代码与GPU实现一样紧密地进行了优化,因此图4显示了不同硬件架构之间的公平比较。
CPU定时基准测试是用Intel Xeon 8375C(Ice Lake)的四个CPU内核进行的,为了简单起见,将结果转换为一个CPU内核的结果。请注意,QSimulate针对CPU硬件的生产平台使用四个CPU内核,与此计时基准测试中一样,主要是因为很难将CPU模拟扩展到更多线程和CPU内核。
 图4。针对CPU内核上的QM/MM FEP模拟的GPU加速
图4。针对CPU内核上的QM/MM FEP模拟的GPU加速
即使是由74个量子力学原子组成的最小量子力学区域,对于A100和H100卡,GPU卡和CPU内核上的量子力学原子的吞吐率分别约为140和170。随着量子力学区域变大,这个比率变得更加显著。对于200个量子力学原子,使用H100卡的吞吐量等于使用265个CPU内核的吞吐量。
人工智能时代的仿真
随着人工智能正在改变社会,基于物理的仿真比以往任何时候都更加重要。复杂工作流的数字转换,如药物发现中的工作流,需要通过准确的模拟方法快速可靠地评估人工智能预测,然后将更多数据输入人工智能模型。本文总结了在毫秒/时间步长范围内首次基于量子力学的模拟的进展,展示了QSimulate的领域知识和软件工程以及NVIDIA硬件和软件,包括最新的CUDA工具包以及软件堆栈,帮助实现了向下一个数字发现范式的飞跃。
要了解更多信息并开始使用GPU加速量子力学模拟进行药物发现,请访问QUELO-G产品页面.