来自Proteopedia
蛋白质组链接蛋白质组链接
核磁共振结构测定
在蛋白质数据库于2019年1月通过溶液核磁共振(NMR)测定。89%由X射线晶体学,2%电子冷冻显微术,其他方法约为1%。核磁共振只能用于相对较小的大分子(参见在下面).
核磁共振波谱是基于自旋为1/2的原子核的能力(例如。1H、,13C、,15N、,31P) 在磁场中采用两个不同的方向。原子核在两种状态之间的分布可以通过使它们受到与它们之间的能量差相称的频率的短脉冲辐射来改变。监测随后衰变中的磁信号可以获得有关原子核方向和间距的动态信息,这些信息提供了可以转化为结构信息的约束[1].
核磁共振分析产生的主要数据主要是关于结构内原子的局部和最近的全局几何信息。通常,这些信息包括原子对之间的距离、二面角(通常是骨架φ角和一些侧链χ1角),有时还包括全局信息,例如给定键相对于分子固定轴的方向。这些数据被用作“约束”,以重建与核磁共振数据兼容的三维模型。所有计算都是直接在物理空间中进行的,从大分子的随机构象开始,大分子逐渐折叠以满足约束条件。通常,从不同的初始构象开始执行多次运行,以检查计算是否收敛到单个解决方案。因此,结果是模型的集合,其分布给出了核磁共振结构精度的度量。
核磁共振实验的模型构建通常从完整的蛋白质或核酸链开始,包括氢原子。然后应用距离限制。结果模型通常包括整个蛋白质和核酸链,不像X射线晶体学由于以下原因,模型通常缺少末端,甚至链条中间也没有回路混乱在蛋白晶体中。
核磁共振法测定大分子结构是在高蛋白浓度的水溶液中进行的,因此要求分子具有高溶解性。有关更多信息,请参阅维基百科中的核磁共振[2]和PDB三维结构数据的性质[3].
显示核磁共振模型
蛋白质组显示核磁共振模型
对于核磁共振实验中的模型集合,Proteopedia最初只显示第一个模型,即通常的卡通渲染;这样做是为了加速页面加载。您将在JSmol面板中看到“Displaying simplified model”消息。如果您点击“加载满”按钮(橙色),Proteopedia将显示所有型号,使您能够看到模型之间的一致之处以及不同之处。每个模型都显示为一条薄的主干线(连接氨基酸的α-碳原子或DNA或RNA链中的磷原子的线)。主干痕迹的颜色为氨基到羧基“彩虹”,一种颜色的光谱序列,从氨基酸末端(或核酸链的5'末端)开始,到羧基末端(或3'末端)结束。
配体(异原子)也显示了所有模型,只是它们仅对模型1是不透明的,而对所有其他模型是半透明的。配体原子按元素着色,使用CPK配色方案具有与链末端共价连接的杂基团且位置极为可变的示例如下1个jsa和1dqc.1巴在可变位置也有杂基。1小时只有杂原子。
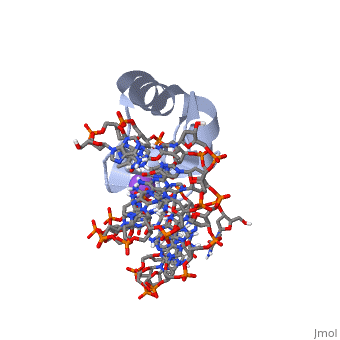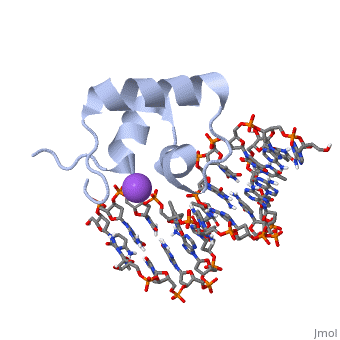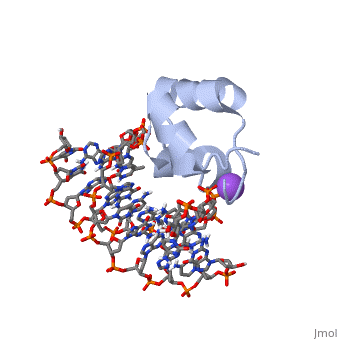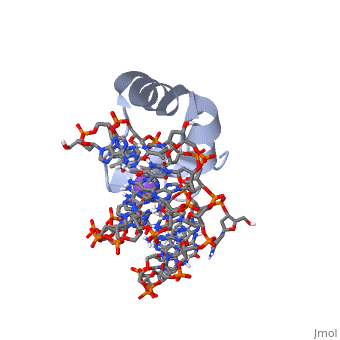
单击“load full”按钮后,右侧的示例显示了1个液晶显示器,一个与DNA结合的紫胶阻遏物结构域,带有一个钠离子。水存在于该模型中,但为了清晰起见,Proteopedia在其初始场景中没有显示水。。(要隐藏水,请单击初始场景分子下方的绿色链接。)
二硫键显示为连接主干的黄色杆,第一个模型不透明,所有其他模型半透明。一个例子是1iw4公司.
单个模型
Proteopedia默认只显示第一个模型,但它说显示简化模型。单击后橙色满载按钮,将显示所有型号。
要查看单个模型,请单击JSmol公司或Jmol_S公司(分子下方的右下角)到打开Jmol的菜单。在这里,使用所有N个型号项目(其中N是集合中模型的总数)。例如,单击1.1:1将仅显示型号1,菜单现在将显示型号1/N。也可以使用Jmol的菜单更改渲染和着色。
Jmol简介默认情况下也显示模型1,但您可以单击查看所有模型.
核磁共振信号群动画制作
当核磁共振集成中的模型像电影一样播放时,生成的动画会模拟热运动(尽管并非所有的运动都是真实的——请参见在下面). 要设置模型的动画,请单击JSmol公司或Jmol_S公司(分子下方的右下角)到打开Jmol的菜单。选择动画,然后动画模式,然后单击循环。然后选择动画再次单击播放。可以使用更改动画的速度FPS(FPS)(每秒帧数)动画菜单。默认情况下,第一个和最后一个模型存在延迟。
核磁共振的多模型集成
核磁共振实验产生多种模型
当通过溶液中的核磁共振(NMR)测定大分子结构时,结果是多分子模型的集合与实验数据一致。核磁共振实验的结果是大量的原子间距离约束,这与多个模型一致。这与X射线晶体学实验的结果相反,该实验是一个最符合经验电子密度的单一模型。(在某些分辨率很高的情况下,模型可能包含一些原子的替代位置。)
发表的核磁共振模型数量取决于实验结果,由作者决定,在2个(例如。1桶)100多个。模型的中位数是20。(您可以使用亚欧理事会). 集合中的第一个模型没有特殊意义(参见最具代表性的模型).
模型之间变化的意义
这个模型之间的差异在合奏中可以指两件事中的任何一件。变化可以代表实际柔韧性和热运动在溶液中进行核磁共振测量时发生的,通常是在室温下。或者,这种变化可以简单地表示原子位置的不确定性也就是说,用于确定某些原子位置的约束数量不足。不幸的是,没有什么可以与B值或温度值这量化了晶体学结果中每个原子位置的不确定性。然而,特定的核磁共振弛豫实验可用于测量单个原子(主要是主链酰胺基)的动力学,因为核磁共振信号的弛豫确实取决于分子的内部运动。当这些核磁共振弛豫数据可用时,它们可用于确定订单参数,与B值晶体结构。这些可以用来区分内在灵活性和由于缺乏约束而产生的不确定性。当弛豫数据不可用时,找出模型之间变化的意义的唯一方法是联系编写已发布模型集合的实验者。
蛋白质链通常在末端的模型之间比中间的模型之间有更多的差异。一个例子是2年.
使用适当的方法,可以确定平均结构及其动态运动[4].
最具代表性的模型
这个最具代表性的模型是最接近平均模型的模型。名为的服务器奥尔德拉多报告最具代表性的模型,并允许您单独下载。
最小平均模型
通常对核磁共振实验中的模型求平均值,但为了使结果真实,必须进行一些能量最小化,以调整共价键的长度和角度。结果称为最小化平均模型有时,作者会同时发布集合和最小平均值。例如2亿立方米似乎是21个模型集合的最小平均值20亿,但如果不阅读原始出版物或联系作者,则很难确定(因为pdb文件不说)。
核磁共振模型的可靠性
核磁共振模型更有可能包含重大错误[5]而晶体学模型分辨率和自由R值。另请参见分子模型的质量评估2012年,完整膜二酰甘油激酶的X射线晶体结构,3季度4,揭示了功能重要的域交换[6][7]早期核磁共振结构中不存在的2千立方厘米[8].
已发表核磁共振结构的中位数
溶液核磁共振无法确定超过30000道尔顿的分子的原子分辨率蛋白质结构。事实上,在蛋白质数据库约为9 kD,90%小于19 kD[9]相反,晶体学确定结构的中间质量为45 kD,其中90%<145 kD。
模型的对齐
核磁共振模型通常是结构上对齐由作者在出版前完成。然而,也有一些例外,例如第1季度6,1dl0个、和1我25其中单个模型未对齐。在这种情况下,需要考虑单个模型为了了解分子结构。
对齐会影响您对模型之间差异的感知。例如,钙调蛋白含有两个EF-手通过灵活的连接器连接。当钙调素不与同源肽结合时,两个EF键可以相对移动,从而弯曲连接子。在1立方英尺,N端EF-手是对齐的,但C端EF手的方向不同。或者,如果C端EF-hands已对齐,则N端EF-hand将处于可变方向。更不可信的是,如果柔性连接器的短中心段对齐,两端将处于可变方向。
通过柔性连接体连接的两个折叠结构域(锌指)的另一个例子是1组1。同样,只能对齐一个域,并且哪个域是任意的。
另请参见
参考资料和网站
- ↑引自该书第22页组件和机器的分子生物学史蒂文、鲍迈斯特、约翰逊和佩勒姆,加兰/CRC出版社,2016.
- ↑ 维基百科中的核磁共振
- ↑ 3D结构数据的性质(存档副本)
- ↑蛋白质结构和动力学的同时测定。Kresten Lindorff-Larsen、Robert B.Best、Mark A.DePristo、Christopher M.Dobson和Michele Vendruscolo(2005年)。自然433:128。PMID:15650731.
- ↑传统的核磁共振波谱生物分子结构测定存在较大误差。Sander B.Nabuurs、Chris.A.E.M.Spronk、Geerten W.Vuister和Gert Vriend。(2006). 公共科学图书馆计算生物学2:Open Access全文 Precis公司DOI:10.1371/journal.pcbi.0020009
- ↑郑洁,贾姿。结构生物学:微小酶利用上下文获得成功。自然。2013年5月23日;497(7450):445-6. doi:10.1038/nature12245。Epub 2013年5月15日。PMID:23676672数字对象标识:http://dx.doi.org/10.1038/nature12245
- ↑Li D、Lyons JA、Pye VE、Vogeley L、Aragao D、Kenyon CP、Shah ST、Doherty C、Aherne M、Caffrey M.完整膜二酰甘油激酶的晶体结构。自然。2013年5月23日;497(7450):521-4. doi:10.1038/nature12179。Epub 2013年5月15日。PMID:23676677数字对象标识:10.1038/自然12179
- ↑Van Horn WD、Kim HJ、Ellis CD、Hadziselimovic A、Sulistijo ES、Karra MD、Tian C、Sonnichsen FD、Sanders CR。膜积分二酰甘油激酶的溶液核磁共振结构。科学。2009年6月26日;324(5935):1726-9. PMID:19556511数字对象标识:324/5935/1726
- ↑在Protein Explorer上:蛋白质数据库中43000个条目的大小和冗余(截至2007年4月)。
外部资源