为了证明QUICS方法准确分离和组织与共洗脱生化物质相关的离子的能力,使用GC/EI-MS分析了33份肝脏样品的样本集。在本研究中,三种已知代谢物,亮氨酸、磷酸盐和甘油,持续共洗脱,如图所示1。如果事先不知道该扫描的内容,则很难确定图中所示的光谱是否1是代谢物的混合物或单个代谢物。使用QUICS方法,通过根据样本集中的仪器响应将高度相关的单个离子分组,将该光谱分为三个生化成分。这是可能的,因为来自单个生物化学的离子在整个研究中会表现出类似的生物可变性,因此相互关联,如图所示2因此,可以基于相关性对属于单个组分的各个离子进行分组。在图中2图A显示了亮氨酸的GC/EI-MS分析过程中产生的两种离子(158和232 m/z)的色谱剖面图。请注意,所示四种不同肝脏样品中离子的强度趋势相同。更具体地说,以黑色显示的肝脏样品的含量最高,分别为158和232 m/z,其次是以红色、绿色和蓝色表示的肝脏样品。因此,当比较研究中所有样品进样的离子响应时,这两种离子相互关联,如图所示三这与甘油相关的离子形成对比(图2,面板B),其中用黑色标记的肝脏样品具有最高数量的两种离子(205和103 m/z),其次是绿色,然后是蓝色,最后是红色。这三种离子群是由一组33个肝脏样品创建的,它们各自的真实标准光谱匹配如图所示4.
迄今为止讨论的示例来自电子电离(EI)GC/MS分析生成的数据,其中检测到的所有离子都是电离期间完整分子碎裂的结果。然而,QUICS方法对其他类型的数据也很有用。去卷积LC/MS数据,其中生物化学化合物不一定在来源中片段化,而是容易形成加合物和多聚体也是可能的。如图所示5,面板A是LC/MS样品注入离子的示例,根据相关性进行分组。呈现的离子是肌苷的各种加合物、同位素、多聚体和源内片段,如B组中真实的标准库光谱所证实。C组中显示的是269 m/z质子化分子离子与137 m/z源内片段之间的相关性。QUICS方法也适用于LC/MS应用,其中从色谱系统中洗脱的所有离子都是碎片化的,而不是分离单个质量然后碎片化。在这种情况下,来自单个代谢物的碎片离子也会根据生物变异性而相互关联。
QUICS方法分离共洗脱物种的能力取决于整个研究中检测到的离子面积响应的相关性。这类分析的一个潜在的复杂因素是,样本集中生物变异性有限的化合物中的离子不会高度相关,因为离子信号的变异有限。此外,相关性计算可能会被共洗脱化合物中共享的离子搞混。在这些情况下,离子响应是所有共洗脱化合物的总和,因此相关性可能会受到影响。图中所示的147和72.9 m/z离子就是这种现象的一个例子1如图中真实的标准光谱所示,这些离子对所有三种共洗脱化合物都是常见的4、面板B、D和F。最终,这些离子与具有足够关联度的化合物分组,特别是与磷酸盐73分组(图4,面板A)和147(图4,面板E);这两种离子相关性都不足以与其他亮氨酸离子分组(图4,图C),尽管亮氨酸标准品也产生了这些离子(图4,面板D)。虽然这些潜在的并发症确实存在,但它们很少会在很大程度上干扰,从而影响所生成频谱的质量。
而图中的示例4重点关注三种已知化合物共同洗脱的色谱区域,当样本集的化学成分未知时,QUICS方法最有效。在这种情况下,即使在化学特性未知且可能与其他未知物质共同洗脱的情况下,也可以对来自每种单独化学物质的单个离子进行分组。如图所示6,面板A是使用QUICS方法创建的频谱的一个示例。分组的离子(图6,A组)来源不明,后来被鉴定为Equol(4',7-异黄素),一种异黄酮,通过肠道细菌群从大豆黄酮代谢而来[9](图6,面板B)。
QUICS方法的最终目标是允许对每个生物化学实体产生的许多冗余离子信号进行反褶积。在生物化学分析领域,数据分析的标准方法之一是使用每个单独的离子信号进行统计分析,无论它们是否是由单个化学物质产生的多余离子[10——12]。由于统计分析中处理的测量数量增加,这种以离子为中心的方法导致了更多的错误发现。由于不同的化学物质会产生不同数量的离子信号,因此这种方法也有可能扭曲统计数据。例如,主成分分析(PCA)等多元技术可能会偏向于产生更多离子信号的化学物质。相反,QUICS方法支持以化学为中心的数据分析方法。一旦组织和分组了属于某一特定生化物质的相关离子特征,该组中的单个离子就可以在统计分析中表示该代谢物。通过使用以化学为中心的方法,减少了错误发现的数量,因为统计分析中处理的离子数量减少为每个代谢物的单个代表性离子,而且,减少了统计结果出现偏差的可能性。
核磁共振数据也使用了类似的跨样品相关性分析。在该分析中,单个分子产生的多个化学位移峰可以跨样品关联、分组,并用于帮助识别检测到的分子[13——16]。虽然在概念上与这里介绍的方法类似,但由于底层数据流是如此独特,因此最终结果具有不同的优势。QUICS方法的目标之一是消除传统上在大多数基于质谱的技术中看到的单个分子产生的高度冗余的离子特征。如前所述,数据的冗余会改变统计分析,并导致更多的错误发现。使用QUICS方法,可以在使用或不使用已知光谱库的情况下,将与检测到的代谢物相关的所有冗余离子分组到一组实验样品中。通过同时利用色谱时域和离子响应,QUICS方法能够获得核磁共振数据流中没有的额外特异性。因此,只对那些共同洗脱和相关的离子进行分组,从而消除了混杂的冗余。应该注意的是,该方法代表了一个自动化软件包,它还能够生成未命名/未知代谢物的光谱库条目,这些分子的化学光谱特征参考库条目尚不存在。采用统计方法分析样品中的所有离子并同时评估整个样品集,使系统不仅能够识别与参考库中已知生化物质光谱匹配的代谢物,还能够识别不在参考库中的生化物质。这些所谓的未命名代谢物的鉴定利用了这样一个事实,即源自单一生物化学物质的离子在研究样本中表现出类似的生物变异性,因此具有相关性。QUICS方法已成功用于多种实验研究,包括疾病生物标记物识别、药物作用模式、毒理学、老化以及复杂混合物(如牛奶)和各种样品基质(如生物流体、组织、牛奶)的变化特征[17——22].
实验
首先,收集GC/MS和LC/MMSS数据,作为自动化、高通量处理系统的一部分。这些数据由离子峰组成,这些离子峰是从每个样品的原始3D GC/MS或LC/MS分析中自动检测和整合的。这些数据的特征是离子的质量(m/z)、面积(代表离子的量)、保留时间(RT)和保留指数(RI),这些色谱特征表示相关生化物质何时洗脱。通过使用添加到每个样品中的内标保留指数及其每个样品的保留时间进行校准,将保留指数分配给所有离子峰[6]。这些原始信号数据和集成离子峰数据均加载并存储在关系数据库系统中,该数据库系统提供了最适合存储和检索大型色谱数据集的数据结构。样本数据采集完成后,系统立即检索数据文件,并完成自动加载和峰值检测,使用匹配算法将所得数据与包含已知标准和未知代谢物光谱定义的现有光谱库进行比较。离子峰数据和光谱库之间发生的任何匹配都会进行评分,以确保可信度,并且这些信息存储在关系数据库系统中。
为了确定样品中哪些离子组是常见的,所有离子都由质量窗口和RI窗口进行装箱。来自同一化学物质的离子被确定为在样品中常见,然后根据相关性进行分组。表1显示了离子箱和箱分组的处理阈值。
离子仓
第一步是开始一个装箱过程,根据质量和保留指数(RI)对同一研究集中样品中的离子进行装箱[23]。bin是一种空间分区数据结构,支持快速区域查询和最近邻搜索。每个箱子的特点是有一个中心质量和一个中心RI。如果来自整个样品组的离子的质量和RI落入中心质量和中心RI周围的窗口中,则将其放入同一箱子中。
在这个过程中可以定义两种离子,一种是单线态,另一种是多重态。单重离子来自同一容器中只有一个离子的样品。多重离子来自于同一容器内有多个离子的样本。来自同一样品的多重离子意味着多个化合物的共同洗脱,称为碰撞。图中显示了加载数据集中的碰撞次数,即在所有箱子中检测到多个离子的次数5(C).
装箱包括以下步骤:
-
1
按面积降序排列离子。
-
2
离子周围面积较小、面积较大的二元离子,较大的离子作为二元中心。
-
三。
计算每个离子仓的统计数据:分别来自仓中所有单线态离子的平均质量、平均面积、平均RI及其标准偏差。
-
4
将料仓中心质量和中心RI重置为其平均质量和平均RI,以考虑料仓内的离子分布。移除所有没有单重离子的料仓。
-
5
将所有离子重新注入这些容器。如果一个离子不能被装箱到其中任何一个中,就会创建一个新的箱子,以其质量和RI为中心质量,RI为。
-
6
重复步骤3和4以优化所有离子的装箱。
离子箱中单线态离子的数量表示该离子在样本中的流行程度。箱子的大小由单重离子在样品总数中的百分比表示,图中的“填充”百分比5(C,%列)。
当垃圾箱未100%装满时,也就是说,当垃圾箱中存在没有离子的样本时,这些样本可能具有相同的离子,但它们可能正好位于垃圾箱的质量窗口和/或RI窗口之外。如果这些异常离子与垃圾箱中的离子相同,它们的面积将在仓中单重离子面积的四个标准偏差内。为了从缺失样品中恢复这些异常离子,在扩展质量窗口和/或RI窗口中搜索仓中“填充”百分比较低的样品;如果找到,这些离子会从“填充”较少的容器迁移到“填充”较多的容器。
例如,假设总共有30个样本,并且有25个样本在仓B1中具有单峰,而样本A中没有任何峰值。为了检查样本A是否具有与仓B1中其他样本的峰值相似的异常峰值,在扩展的RI和质量窗口中搜索相邻仓中样本A的峰值。样品A的峰面积必须在B1中峰面积的四个标准偏差窗口中。相邻料仓中样品A的最佳匹配峰将迁移到料仓B1,使料仓B1更“充满”(现在包括样品A)。
此过程从装满较多的箱子循环到装满较少的箱子。
分组离子仓
在GC/MS或LC/MS中,在电离和裂解过程中,同一代谢物可能会产生许多离子。在LC/MS中,同一代谢物可形成不同的加合物和聚集体。来自同一代谢物的这些离子应具有良好的相关性。另一方面,来自不同化学来源的离子在很大程度上是非协变的。
一旦样本集中的离子被正确地装箱,每个箱子代表一个在样本集中许多样本中常见的公共离子。在对多个样品进行分析时,代表同一代谢物离子的Bin应具有良好的相关性。假设大多数样品都含有一种常见的代谢物a,该代谢物已电离为N个离子,那么就会有N个相互关联的垃圾箱。对垃圾箱进行分组的目的是找到那些相关性很好的离子,这些离子可能代表这些样本中已知或未知的代谢物。
归一化箱和相关箱之间的相关性
计算皮尔逊相关性以测量两个离子箱之间的相关性。计算中只包括两个离子箱中常见的单重离子。
箱子按平均面积降序排序。使用较大的容器作为标准化器,较小的容器围绕较大的容器进行分组,就像相关性高于相关阈值一样。归一化器bin和相关bin之间的相关性计算如下:
其中S我是普通单线态离子的面积,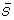 是一个箱子中常见单重离子的平均面积。最小相关阈值和归一化箱子和相关箱子中常见的单重离子数量由用户指定,可以在进程启动之前更改。
是一个箱子中常见单重离子的平均面积。最小相关阈值和归一化箱子和相关箱子中常见的单重离子数量由用户指定,可以在进程启动之前更改。
相关阈值通过试验和误差选择。通常在0.70到0.90之间。它取决于矩阵类型和样本集大小(样本集中的样本数)。相关性阈值过低会将过多的垃圾箱分组到一个组中,而阈值过高则会遗漏组中的一些离子,这可以从库匹配中已知样本的代谢物中判断。
化学情报
源自同一化学物质的离子将具有不同的m/z值,并包括各种源内碎片、同位素、加合物和多聚体。在LC/MS中,离子可以是单体或与溶剂/流动相离子(如H)的加合物的聚集体+,纳+,K+,氯-,俄亥俄州-,新罕布什尔州4+,H2O、 首席运营官-等。可根据测量质量计算真实质量:
其中N输入法是骨料中单体的数量,(m/z)加合物是加合物的(m/z)单体,是单体的(m/z),以及仔细斟酌的是离子的测量值(m/z)。
表2显示了最常见的聚集体和加合物。根据这些可能的聚集物和/或加合物检查组中的所有离子,以确定离子的最可能形式。
为了计算代谢物的单体质量,根据上述可能的离子形式对分组箱中的每个离子进行测试,并通过离子峰面积和离子形式概率的乘积计算和评分可能的单体重量。
为此,计算所有离子形式的所有离子的可能单体质量,并在表中进行分类和评分三:
1.适用于每个离子AND组从大(大离子峰面积)到小(小离子峰面积每种离子形式从可能性最大到可能性较小的离子形式
2.在所有可能的单体质量中,质量分数最大的单体是最可能的质量。
从组中关联良好的容器中计算单体质量后,可以搜索组中容器中不存在的来自相同代谢物的其他形式的加合物/聚集离子。这些缺失形式的加合物/聚集物离子在样品中可能会有更大的差异,因此它们与归一化bin的相关性低于用于分组的阈值,因此代表这些加合物或聚集物的bin被错误分组为不同的组。QUICS方法试图通过降低质量与代谢物单体质量一致的离子所需的相关阈值来纠正这些分组错误的离子。
对于每种可能形式的加合物/集料,要在一个较大的组(归一化池的峰面积较大)中找到缺失的加合产物/集料并按照上述方法计算其单体质量:
-
1
假设存在这种加合物/聚集体形式的离子,根据单体质量计算测量质量。
-
2
在计算质量的质量窗口(±0.4)和较大组归一化箱的RI窗口内搜索较小组中的箱。
-
三。
计算这些加合物/聚集物箱与大组归一化箱的相关性。
-
4
相关性最高超过0.4的离子箱将是缺失的加合物/聚集离子之一,并将从较小的组迁移到较大的组。
每个小组从大到小重复这个过程。
同位素离子的检查方法相同,只是要求离子峰面积不超过正常离子的一半。
如上所述,每个主要离子峰位代表样品中一个普通离子的平均值,而每组相关的位代表一种普通代谢物的离子。对于LC/MS/MS,每个一级离子的二级MS2离子也可从一级离子峰仓和类似仓中的所有单重态样品中检索。在满足最小单重离子数的MS2离子仓中,平均强度最大的最大仓将用作归一化仓,以归一化其他既满足最小单轻离子数又满足最小相对强度的仓,包含在库中以表示主离子的MS2离子。总之,根据保留指数和离子质量的色谱特征,对所有样品的综合初级离子色谱峰进行了分类,并对保留指数窗口内相关良好的分类箱进行了分组,以创建一个表示化学实体的库条目。每个质量及其特征值来自组中一个仓的质量、面积、RI和RT的平均值,代表来自纯化学实体或其加合物/聚集体的碎片离子之一。对于LC/MS/MS,样品中所有二次离子的桶/组也会产生二次质量,平均值代表化学实体一个主要碎片的二次离子。为每个化学实体创建的此类库条目可能与库中的条目匹配良好,或者作为未知条目添加到库中,并通过更多研究进一步确定。
参考库条目
一旦根据相关性对装箱离子进行分组,就可以根据参考库搜索组,以确定离子组是否代表已知实体,或者离子组是否表示新的或未知的(库中不存在的生物化学)化学实体。如果离子组被确定为代表一种没有参考库条目的未知化学物质,则会在库中添加一个新的光谱条目,以便在未来的研究中跟踪未知实体。试图根据先前定义的质量关系,例如Na加合物m+23,将化学情报分配给属于未知实体的离子。








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}